有机核心考点
有机物概述
同分异构现象
化合物具有相同的分子式,但具有不同结构的现象,称为同分异构现象。具有这种现象的化合物互称为同分异构体。
碳链异构:碳链骨架不同。
\ce{CH3CH2CH2CH3} 和 \ce{CH3CH(CH3)2}
位置异构:官能团或取代基在碳骨架(碳链或碳环)上位置不同。
\ce{CH2=CHCH2CH3} 和 \ce{CH3CH=CHCH3}
\ce{CH3CH2CH2OH} 和 \ce{CH3CH(OH)CH3}
\ce{CH3OCH2CH2CH3} 和 \ce{CH3CH2OCH2CH3}
\ce{R1COOR2} 和 \ce{R2COOR1} (R1 \neq R2,且均为烃基)
官能团异构:官能团不同。
\ce{CH3CH2OH} 和 \ce{CH3OCH3}
\ce{CH3CH2CHO} 和 \ce{CH3COCH3}
\ce{CH3COOH} 和 \ce{HCOOCH3}
顺反异构:原子或原子团在碳碳双键上的位置不同。
从复杂基团到简单的为正方向,当两侧均为同一方向,为顺,反之为反
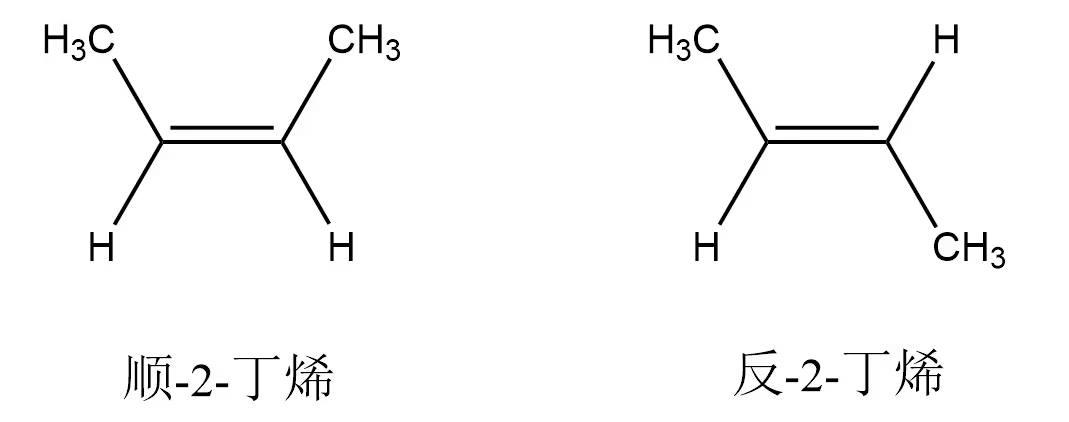
双键上的碳原子及与其直接相连的原子位于同一平面,碳碳双里中任意一个双键碳原子上连接 2 个相同的原子或原子团时,不存在顺反异构
对映异构:互为镜像且不能重叠的结构,即存在手性碳原子便存在对映异构。
手性碳原子:饱和碳的周围接了 4 个两两不同的原子或原子团

注意:题目中如问给定结构的物质的同分异构体数目,需要扣除该物质本身。
原子共面问题
可能出现的题目要求:
「碳原子」、「所有原子」。
「一定」、「可能」、「至少」、「最多」。
「共线」、「共面」。
选定主体结构:
凡出现苯环结构形式的原子共面问题,以苯的结构为主体
凡出现碳碳双键、碳碳三键结构形式的原子共面问题,以乙烯、乙炔的结构为主体。
画出有机化合物的结构式,观察原子的共线、共面情况。
单键可以旋转,双键、三键不可以旋转。
注意:
- 结构中出现饱和原子,则不可能所有原子共平面。
- 结构中每出现一个碳碳双键,至少有 6 个原子共面。
- 结构中每出现一个碳碳三键,至少有 4 个原子共线。
- 结构中每出现一个苯环,至少有 12 个原子共面。
烷烃异构问题
烷烃的同分异构书写常用减碳法。
三注意:注意要选择最长的碳链作主链;注意要找出对称轴;注意要保证每次减掉碳原子后的碳链仍为主链。
三原则:对称性原则、有序性原则、互补性原则。
四顺序:
- 主链由长到短:选取最长的碳链为主链,再逐步减少主链的碳原子数,余下的碳原子作为取代基。
- 取代基由整到散余下的碳原子先作为一个取代基,再逐步拆散为多个小取代基。当有多个取代基存在时,应按连接在同一碳原子、 相邻碳原子、相间碳原子…的顺序依次移动,避免漏项
- 位置由心到边不到端:把取代基连在主链上,由主链的对称中心开始,逐步向一边移动,但注意不要移到端点
- 排列由对、邻到间:两个取代基可以相对 (连在同一个碳原子上)、相邻(分别连在相邻的两个碳原子上)和相间(分别连在不相邻的 两个碳原子上)。
注意:利用减碳法书写同分异构体时应注意保证减掉碳原子后的碳链仍为主链,如甲基连在主链的端点碳原子上、乙基连在主链 的二号碳原子上,均会改变主链,导致书写重复。
具有官能团的有机化合物同分异构体的书写:书写步骤为先确定可能含有的官能团类别,之后按照烷烃同分异构体的书写方法分别写出除官能团外的碳链异构,然后再移动官能团的位 置,最后按照碳原子形成四个共价键的原则,把氢原子补齐。
烷烃的同分异构计数没参考 OEIS A000602 给出下面的几组,详见 Luogu P6598 烷烃计数。
| \ce{CH4} | \ce{C2H6} | \ce{C3H8} | \ce{C4H10} | \ce{C5H12} | \ce{C6H14} |
|---|---|---|---|---|---|
| 1 | 1 | 1 | 2 | 3 | 5 |
烷基的同分异构计数参考 OEIS A000598 给出下面的几组,详见 LOJ 6538 烷基计数。
| \ce{-CH3} | \ce{-C2H5} | \ce{-C3H7} | \ce{-C4H9} | \ce{-C5H11} | \ce{-C6H13} |
|---|---|---|---|---|---|
| 1 | 1 | 2 | 4 | 8 | 17 |
一元取代计数
等效氢法:
有机化合物分子中有几种不同化学环境的氢原子,则其一元取代物就有几种同分异构体。
- 同一碳原子上的氢原子是等效的,如 \ce{CH4} 分子中的 4 个氢原子是等效的。
- 同一碳原子所连的相同基团上的氢原子是等效的,如新戊烷分子中的 4 个甲基等效,各甲基上的氢原子完全等效,即新戊烷分子中的 12 个氢原子是等效的。
- 处于镜面对称位置上的氢原子是等效的,如丁烷分子中有种 2 等效氢原子。
我们常在结构式中,用 1,2,3 来标记等效的氢,以此计数。
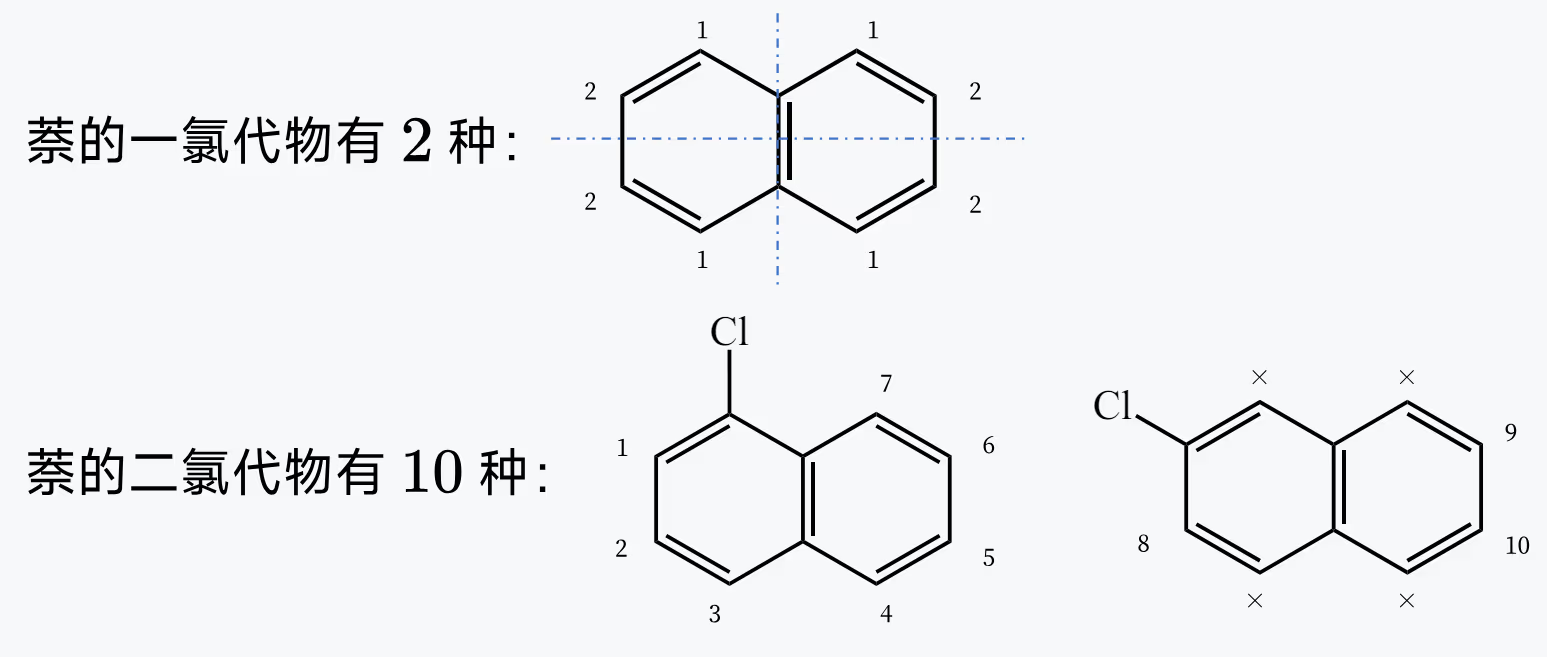
二元取代计数
确定链状烷烃二取代物的同分异构体数目时,可首先固定一个取代基,再按照顺序移动另一个取代基以确定同分异构体数目,下面以确定 \ce{CH3CH2CH3} 的二氯代物的数目为例
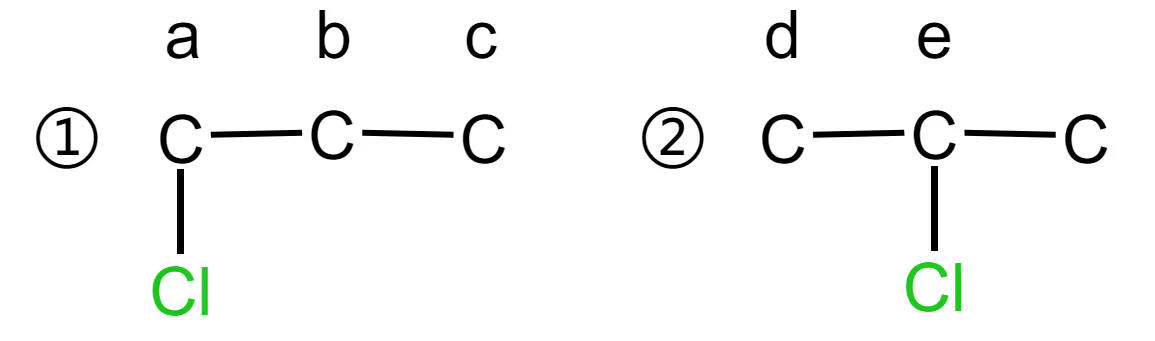
先固定 1 个氯原子,有 2 种(如上图 ① 和 ②)。
然后移动第 2 个氯原子:
- 结构 ① 中有 3 种(在 a, b, c 位置取代)。
- 结构 ② 中有 2 种(在 d, e 位置取代)。
- 其中,在 ① 的 b 位置取代与在 ② 的 d 位置取代后得到的产物(1,2-二氯丙烷)重复。
- 故 \ce{CH3CH2CH3} 的二氯代物共有 3 + 2 - 1 = 4 种。
通过上述案例可以得到:
- 形如 \ce{C3X2} 的二取代物有 4 种。
- 形如 \ce{C3XY} 的二取代物有 5 种。
- 形如 \ce{C4X2} 的二取代物有 9 种。
- 形如 \ce{C4XY} 的二取代物有 12 种。
确定苯环上的二、三取代物的同分异构体数目时(不考虑基团异构),也可以采用类似定一移一法的方法,可以得到:
- 苯环上二取代 \ce{C6H4X2} 的同分异构体数目有 3 种。
- 苯环上二取代 \ce{C6H4XY} 的同分异构体数目有 3 种。
- 苯环上三取代 \ce{C6H3X3} 的同分异构体数目有 3 种。
- 苯环上三取代 \ce{C6H3X2Y} 的同分异构体数目有 6 种。
- 苯环上三取代 \ce{C6H3XYZ} 的同分异构体数目有 10 种。
如苯环上有多个相同的取代基,可将其视为 \ce{H},移动其他取代基计算同分异构体。
烷基取代计数
将有机物分子拆分为烃基和官能团两部分,根据烃基异构体的数目,确定有机物分子的数目。如分子式为 \ce{C4H10O} 属于醇的同分异构体,可改写成 \ce{C4H9—OH},共有 4 种结构;分子式为 \ce{C5H10O} 属于醛的同分异构体,可改写成 \ce{C4H9-CHO},共有 4 种结构。
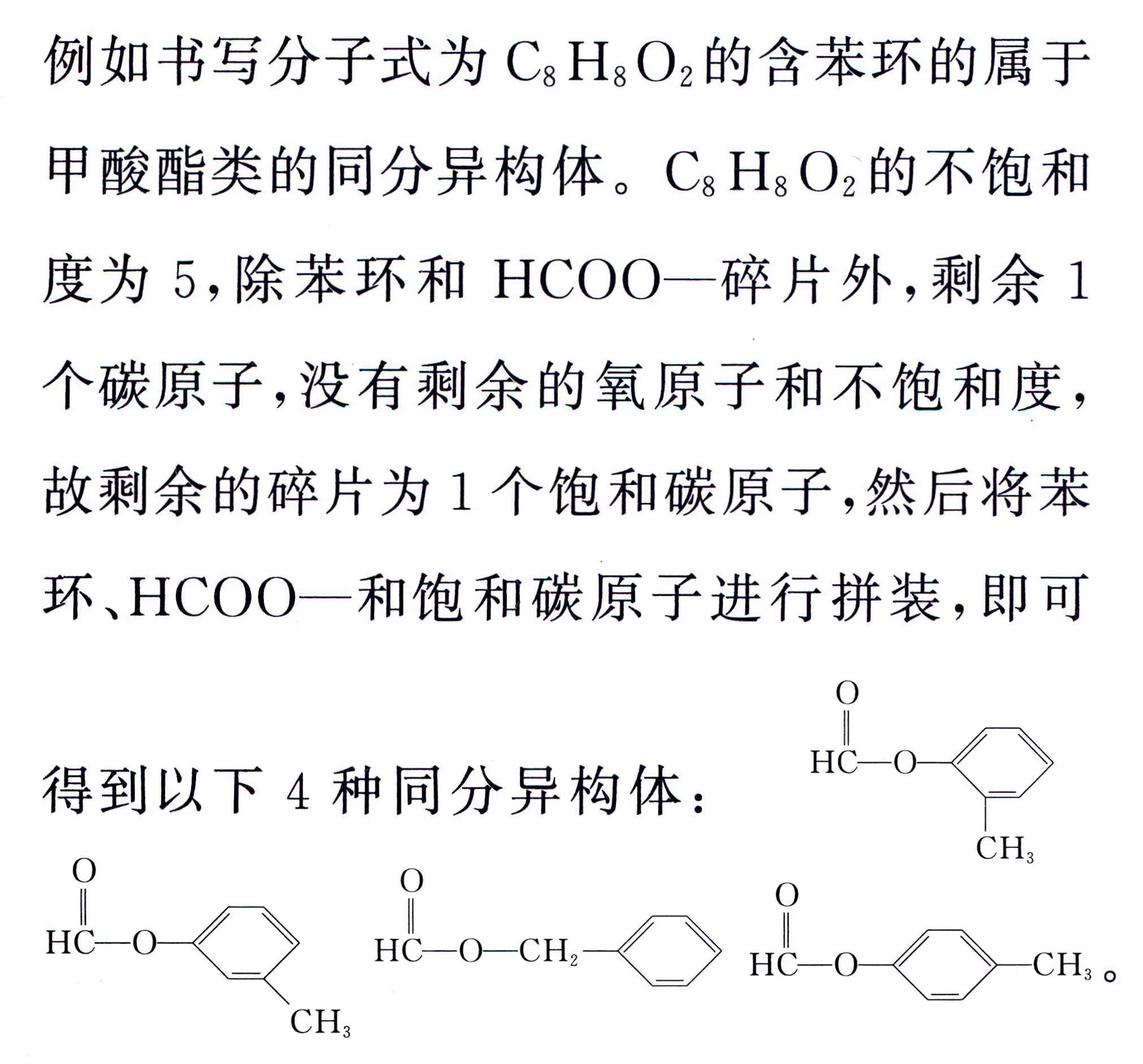
碎片拼接法:

马氏规则概述
马尔科夫尼科夫规则指出:当不对称烯烃与含氢的化合物(\ce{H2},\ce{HCl} 等)加成时,氢原子主要加到连有较多氢原子的碳原子上。
很多反应与马氏规则相悖,这些反应称为反马尔科夫尼科夫规则的反应,例如在过氧化物存在的情况下,氢原子主要加在连有较少氢原子的碳原子上。
具体而言,当 \ce{HBr} 与不对称烯烃反应时,若体系中存在过氧化物(如 \ce{ROOR}),反应机理由离子型亲电加成转变为自由基加成:过氧化物在光或热下均裂产生自由基,引发 \ce{HBr} 产生溴自由基(\ce{Br\cdot}),溴自由基先加到双键上,为生成更稳定的碳自由基(叔 > 仲 > 伯),溴被迫加在含氢较多的碳上,随后该碳自由基从另一分子 \ce{HBr} 中夺取氢原子,同时再生溴自由基以延续链反应,从而得到反马氏产物。例如丙烯在过氧化物存在下与 \ce{HBr} 反应,主要生成 1-溴丙烷而非马氏规则预测的 2-溴丙烷。值得注意的是,过氧化物效应仅对 \ce{HBr} 显著,\ce{HCl} 和 \ce{HI} 因键能差异不表现此效应。
与其相反的,查依采夫规则指出:醇或卤代烃在进行消除反应时,羟基优先与相邻连有氢原子较少的碳原子上的氢原子脱水。
这个规则可以概括为「富者愈富,而穷者愈穷」的马太效应,简记为劫贫济富规则。
需要了解的是,当使用体积庞大的碱(如叔丁醇钾 \ce{(CH3)3CO^-K^+})或离去基团体积很大(如季铵盐 \ce{-N^+(CH3)3})时,查依采夫规则可能失效。大体积碱难以接近分子内部位阻较大的 \beta-\ce{H},会优先夺取分子外围位阻较小的 \beta-\ce{H},从而得到取代基较少的烯烃(霍夫曼产物)。这属于动力学控制——空间位阻效应压倒了产物热力学稳定性。
有机化学实验
乙烯的制取
反应原理:
\ce{CH3CH2OH ->[浓硫酸][\pu{170^oC}] CH2=CH2 ^ + H2O}
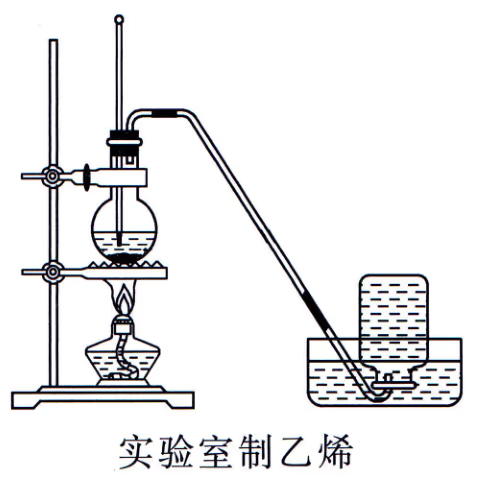
乙醇和浓硫酸的体积比为 1:3,加入沸石或碎瓷片防止暴沸。
浓硫酸起催化剂和脱水剂的作用,制得的乙烯中可能混有 \ce{CO2},\ce{SO2} 等。
为了除去产品中的上述杂志和挥发的乙醇,可用 \ce{NaOH} 溶液除去。
乙炔的制取
反应原理:
\ce{CaC2 + 2H2O -> CH#CH + Ca(OH)2}
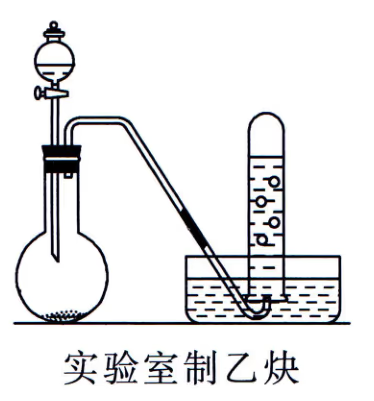
碳化钙是电石的主要成分,反应后可能有 \ce{H2S},\ce{PH3} 等杂志,可用 \ce{CuSO4} 溶液除去。
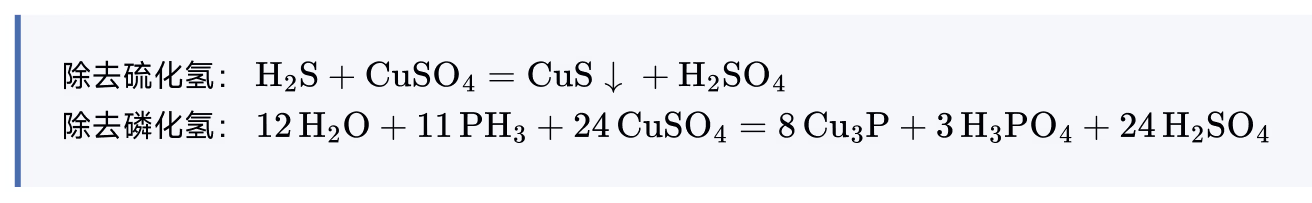
制取乙炔不能使用启普发生器 或具有启普发生器原理的装置,原因如下:
- 碳化钙与水反应剧烈,不能随时停止;
- 反应过程中放出大量的热,易使得启普发生器炸裂;
- 生成的氢氧化钙呈糊状,易堵塞球形漏斗。
为了得到平稳的乙炔气流,常用饱和食盐水代替水,用分液漏斗控制滴加速度。
碳化物水解与有机产物
碳化钙(\ce{CaC2})水解制取乙炔,属于无机化学中类盐水解的范畴。碳化物的水解产物取决于碳阴离子的骨架结构,骨架不变,水解时只加氢,不断键。
\ce{C2^2-}(乙炔型):两个碳以三键相连,水解生成乙炔 \ce{C2H2}。代表物:\ce{CaC2}、\ce{BaC2}。
\ce{C^4-}(甲烷型):孤立的单碳阴离子,水解生成甲烷 \ce{CH4}。代表物:\ce{Al4C3}、\ce{Be2C}。
\ce{Al4C3 + 12H2O -> 4Al(OH)3 v + 3CH4 ^}
\ce{C3^4-}(丙二烯型):三个碳原子累积双键骨架,水解生成丙二烯 \ce{C3H4}。代表物:\ce{Mg2C3}。
\ce{Mg2C3 + 4H2O -> 2Mg(OH)2 v + C3H4 ^}
这条规律可以帮助我们从无机碳化物直接推断有机产物。
乙炔的催化氢化
乙炔(\ce{C2H2})通过选择性催化氢化,可以制备乙烯(\ce{C2H4}):
\ce{CH#CH + H2 ->[Lindlar 催化剂] CH2=CH2}
Lindlar 催化剂(\ce{Pd/CaCO3} + 醋酸铅 + 喹啉)是常用的催化剂,其选择性来源于醋酸铅在 \ce{Pd} 表面形成铅合金,降低钯的氢化活性,从而阻止乙烯进一步氢化为乙烷。
该路线与乙醇脱水制乙烯相比:
优点:过程完整(从无机盐到有机烃),无强酸废液,产气较纯净。
缺点:需要氢气源和催化剂,操作相对复杂。
实验室制备乙烯的常规方法仍然是乙醇在浓硫酸催化下 \pu{170^oC} 消去脱水,而碳化钙水解则主要用于制备乙炔本身。
溴苯的制取
反应原理:
\ce{C6H6 + \underset{液态溴}{Br2} ->[FeBr3] C6H5Br + HBr}
把苯和少量液态溴放在烧瓶里,加入少量铁屑,铁屑与溴反应生成 \ce{FeBr3} 作为催化剂。
常温下,反应生成的溴化氢遇水蒸气生成白雾,烧瓶底部产生溴苯,溴苯的密度比水大、且不溶于水,溴溶解在溴苯中,使溴苯呈现褐色。
不能通过将产物气体通入 \ce{AgNO3} 溶液产生淡黄色沉淀来判断反应的发生,因为产物中含有杂质 \ce{Br2} 蒸气(可用 \ce{CCl4} 洗气)。
苯的硝化反应
反应原理:
\ce{C6H6 + \underset{HNO3}{HO-NO2} ->[浓硫酸][50\sim60\pu{^oC}] C6H5NO2 + H2O}
在试管内先加入少量浓硝酸和浓硫酸,摇匀并冷却到 50\sim60\pu{^oC} 左右,在 60\pu{^oC} 的水浴中加热,慢慢滴加少量苯,继续加热可以看到有一种油状物产生,即硝基苯 \ce{C6H5NO2}。
硝基苯是一种没有颜色的油状液体,有苦杏仁味,密度比水大,难溶于水,易溶于乙醇和乙醚,有毒。
\ce{PhNO2 + 3Fe + 6HCl -> PhNH2 + 3FeCl2 + 2H2O}
硝基苯可以被还原为苯胺,苯胺是重要的工业原料。
乙酸乙酯的制取
反应原理:
\ce{CH3COOH + HOCH2CH3 <=>[浓硫酸][\triangle] CH3COOCH2CH3 + H2O}
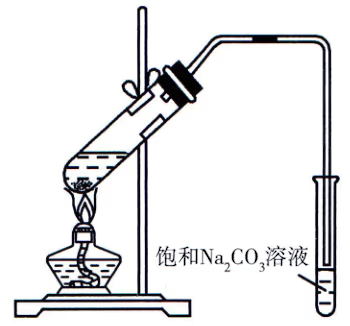
- 试剂的加入顺序为乙醇、浓硫酸和冰醋酸(乙酸),不能先加浓硫酸,浓硫酸作催化剂、吸水剂。
- 导气管不能插入饱和碳酸钠溶液中,防止倒吸,饱和碳酸钠溶液的作用:吸收乙醇、中和乙酸、降低乙酸乙酯的溶解度。
有机推理推断
反应类型总结
取代反应:有机物分子里的某些原子或原子团被其他原子或原子团所替代的反应。
卤代反应:
以烷烃为代表的 \text{sp3} 杂化的碳上的氢原子的光卤代。
苯与甲苯的卤代:
苯和液态溴在三溴化铁催化下可以发生反应,生成溴苯。
在三氯化铁的催化下,甲苯与氯反应生成邻氯甲苯或对氯甲苯(光照情况下甲基发生光卤代)。
苯酚稀溶液的试管里逐滴加入过量饱和的溴水,产生白色沉淀。
醇与浓的氢卤酸,生成卤代烃和水。
硝化反应与磺化反应:
苯与浓硝酸在浓硫酸催化下反应生成硝基苯。
笨与浓硫酸反应生成苯磺酸。
甲苯与浓硝酸在浓硫酸催化下反应主要生成三硝基甲苯(甲基的邻对位)。
脱水反应与水解反应:
醇分子间脱水成醚:\ce{R-OH + HO-R' -> R-O-R'}。
酯化反应与合成酰胺反应:酸脱羟基,醇 / 氨去氢。
需要注意的是,上述结论适用于大多数伯醇和仲醇的酯化反应(遵循 A_{Ac}2 酰氧键断裂机理)。但当醇为叔醇(如叔丁醇 \ce{(CH3)3C-OH})或容易形成稳定碳正离子的苄醇、烯丙醇时,酯化反应的机理发生反转,变为 A_{Al}1 烷氧键断裂机理:醇先质子化后脱水生成稳定的碳正离子,\ce{^{18}O} 以水的形式脱去,随后羧酸作为亲核试剂结合碳正离子。因此,当标记了 \ce{^{18}O} 的叔醇与羧酸反应时,产物酯中不保留同位素标记,\ce{^{18}O} 出现在水中而非酯中,表现为「酸脱氢,醇脱羟基」。
酯类的水解:在酸或碱催化的条件下,酯可以发生水解反应生成相应的酸和醇。酯的水解反应是酯化反应的逆反应。在碱性条件下,化学平衡正向移动,使酯的水解程度加大。
油脂的碱性水解又称为皂化反应。酰胺的水解,如果是碱性水解则生成氨气。
卤代烃在水溶液中与氢氧化钠反应生成醇和卤化钠(注意在醇溶液中发生消去反应生成水和烯烃)。
多糖、蛋白质等大分子有机物的脱水与水解。
酯交换反应:
酯交换反应(Transesterification)是有机化学中一类重要的反应,其本质是醇(\ce{R''OH})作为亲核试剂进攻酯(\ce{R'COOR'})的羰基碳,生成新的酯(\ce{R'COOR''})和新的醇(\ce{R'OH})。
从反应类型看,酯交换属于亲核酰基取代反应,更准确地说,它是酯的醇解反应。从表观结果看,一个烷氧基(\ce{-OR'})被另一个烷氧基(\ce{-OR''})取代,因此定性为取代反应是正确的。
酯交换反应的机理遵循典型的加成-消除两步过程,需要酸或碱催化才能顺利进行。
碱催化机理:在碱性条件下,催化剂通常是醇盐(\ce{R''O^-},由目标醇与强碱反应生成)。第一步,醇盐负离子作为强亲核试剂攻击酯分子中带有部分正电荷的羰基碳,碳氧双键断裂,形成一个带负电荷的四面体中间体。第二步,四面体中间体不稳定,氧原子上的负电荷重新形成碳氧双键,同时将原来的烷氧基(\ce{-OR'})作为离去基团踢出,生成新的酯和新的醇盐负离子。
酸催化机理:在酸性条件下,催化剂是质子(\ce{H^+})。第一步,质子结合到酯的羰基氧原子上,使羰基碳的正电性大大增强。第二步,中性的醇分子攻击被活化的羰基碳,形成带正电荷的四面体中间体。第三步,四面体中间体内发生质子转移,质子从刚加进来的醇氧原子上转移到原来的烷氧基的氧原子上,使难以离去的烷氧基变成容易离去的醇分子。第四步,羟基氧孤对电子重新形成碳氧双键,同时将质子化了的离去基团脱除。第五步,生成的新酯脱去质子,恢复为中性,同时再生酸催化剂。
需要注意的是,酯交换是一个可逆反应。在实际操作中,通常加入大大过量的反应物醇,或将生成的醇(如甲醇、乙醇,通常沸点较低)蒸馏分离出去,利用勒夏特列原理促使化学平衡向正反应方向移动。
加成反应:有机物分子中的不饱和键两端的两个原子与其他原子或原子团直接结合,生成新的化合物的反应叫做加成反应。
碳碳双键、三键等 \pi 键、离域大 \pi 键:烯烃、炔烃、苯的加成。
碳氧双键(醛与酮)的加成:生成醇,为经典的还原反应。
碳氧双键的亲核加成(以醛与氢氰酸为例):
乙醛与氢氰酸发生加成反应生成 2-羟基丙腈,这是一个典型的亲核加成反应。
关于这一反应的两个常见说法:
醛基中的碳原子带部分正电荷,与 \ce{CN^-} 作用:这种说法是正确的。由于氧的电负性大于碳,\ce{C=O} 双键的电子云偏向氧原子,碳原子带有部分正电荷(\delta^{+}),这使得碳原子具有亲电性,能够被带负电荷的 \ce{CN^-} 攻击,形成碳碳键。这在化学上称为亲核加成反应。
\ce{HCN} 有挥发性且有剧毒,是该反应不在酸性环境下进行的原因之一:这种说法也是正确的。原因主要有两点:一是 \ce{HCN} 的沸点只有 25.6\pu{^oC},在室温下极易挥发;二是酸性条件下,平衡 \ce{HCN \rightleftharpoons H^+ + CN^-} 极度向左移动,\ce{CN^-} 浓度极低,反应根本无法进行。因此,该反应通常在弱碱性条件下进行,以增加 \ce{CN^-} 的浓度,提高反应速率。
油脂的氢化或硬化:油脂生成脂肪。
注意烯醇与醛(例如乙醛、乙烯醇)的互变异构。
聚合反应:详见高分子。
缩聚反应:通过取代反应实现的聚合反应。
加聚反应:通过加成反应实现的聚合反应。
逆合成分析与合成路线设计
逆合成分析(Retrosynthetic Analysis)是有机合成路线设计的核心方法,通过从目标分子出发,逐步逆向推导可用的前体和反应,最终得到切实可行的合成路线。
逆推法基本策略
逆推法的核心是切断(Disconnection)——在目标分子中找到合适的键进行切断,将分子拆解为更简单的片段。常用的切断策略包括:
- 官能团转化(Functional Group Interconversion, FGI):将一种官能团转化为它的前体。
- 键的切断:在官能团附近或碳骨架的关键位置切断,暴露可用的反应位点。
- 重排分析:考虑通过已知重排反应构建目标骨架。
苯甲酸苯甲酯的合成路线
苯甲酸苯甲酯(\ce{PhCOOCH2Ph})是一种芳香酯,以下通过逆推法设计多种合成路线。
逆推分析:酯类最经典的切断方式是切断酰氧键(C–O 键)。
\ce{PhCOOCH2Ph => PhCOOH + PhCH2OH}
由此,合成任务转化为如何获取苯甲酸和苯甲醇。高中有机化学中,最基础的起始原料通常是甲苯(\ce{Ph-CH3})。
路线一:经典高中路线(氧化与取代)
- 甲苯 \rightarrow 苯甲酸:\ce{Ph-CH3 ->[KMnO4/H+][\triangle] Ph-COOH}
- 甲苯 \rightarrow 苯甲醇:\ce{Ph-CH3 ->[Cl2/h\nu] Ph-CH2Cl ->[NaOH/H2O][\triangle] Ph-CH2OH}
- 酯化反应:\ce{Ph-COOH + Ph-CH2OH ->[浓 H2SO4][\triangle] Ph-COOCH2Ph + H2O}
Fischer 酯化机理(加成-消除机制):
- 质子化:羰基氧被 \ce{H+} 质子化,增强羰基碳的正电性。
- 亲核加成:苯甲醇的氧原子进攻质子化的羰基碳,形成四面体中间体。
- 质子转移:羟基变成离去能力极强的 \ce{-OH2+}。
- 消除:脱去一分子水,重新形成碳氧双键。
- 去质子化:得到最终产物。
该反应是完全可逆的,实际操作中通常通过分水器除水来推动反应向正方向进行。
路线二:坎尼扎罗反应(Cannizzaro Reaction)
- 甲苯 \rightarrow 苯甲醛:\ce{Ph-CH3 ->[Etard 反应] Ph-CHO}
- 歧化反应:\ce{2Ph-CHO ->[浓 NaOH][\triangle] Ph-COONa + Ph-CH2OH}
- 酸化与酯化:分离得到苯甲酸和苯甲醇后进行酯化。
Cannizzaro 反应机理:
- 亲核加成:\ce{OH-} 进攻苯甲醛的羰基碳,生成负离子四面体中间体。
- 负氢转移(决速步):该中间体上的负氢(\ce{H-})进攻第二分子苯甲醛的羰基碳。
- 质子交换:生成苯甲酸负离子和苯甲醇。
路线三:酰氯法(实验室高产率路线)
- 原料准备同路线一,得到苯甲酸和苯甲醇。
- 制备酰氯:\ce{Ph-COOH ->[SOCl2][\triangle] Ph-COCl + SO2 ^ + HCl ^}
- 酯化:\ce{Ph-COCl + Ph-CH2OH ->[吡啶] Ph-COOCH2Ph}
亲核酰基取代机理:
- 苯甲醇的氧原子进攻苯甲酰氯的羰基碳,生成 sp^3 四面体中间体。
- 四面体中间体崩解,\ce{Cl-} 作为离去基团被踢出,重新形成碳氧双键。
- 吡啶吸收反应产生的 \ce{HCl},拉动反应彻底完成。
路线四:季先科反应(Tishchenko Reaction)——最优雅的一步法
- 甲苯 \rightarrow 苯甲醛(同路线二)。
- Tishchenko 偶联:\ce{2Ph-CHO ->[Al(O-i-Pr)3] Ph-COOCH2Ph}
Tishchenko 反应机理:
- 醇铝催化剂的烷氧基(\ce{RO-})进攻第一分子苯甲醛的羰基,生成半缩醛负离子络合物。
- 该络合物配位第二分子苯甲醛,形成六元环状过渡态。
- 在六元环内发生同步的负氢转移。
- 碳氧键重排,直接生成苯甲酸苯甲酯,同时催化剂脱落完成催化循环。
该反应原子经济性达 100\%,是绿色化学的典范。
其他可行路线
- 酸酐法:苯甲酸脱水生成苯甲酸酐,再与苯甲醇反应。
- 苄基卤与苯甲酸盐亲核取代:\ce{PhCOONa + PhCH2Cl -> Ph-COOCH2Ph + NaCl}
- 酯交换法:\ce{PhCOOCH3 + PhCH2OH ->[酸或碱] Ph-COOCH2Ph + CH3OH}
路线选择建议
- 高中考试:选择路线一,考察高锰酸钾氧化、自由基取代、卤代烃水解和 Fischer 酯化四个核心考点。
- 强基计划、竞赛或面试:选择路线二(Cannizzaro)或路线三(酰氯法),展示对反应产率和中间体利用率的思考。
- 大学有机化学讨论:选择路线四(Tishchenko),展示对原子经济性和负氢转移机理的深刻理解。
消去反应:以醇为代表的羟基 +\beta-\ce{H},马氏规则。
氧化反应:脱氢加氧为氧化。
燃烧。
醇的催化氧化:羟氢 +\alpha-\ce{H}。
醛到酸的氧化,注意甲醛有两个醛基。
溴水、液态溴、酸性高锰酸钾溶液详见试剂。
还原反应:加氢脱氧为还原。
硼氢化钠具有较强的还原选择性,它可以将羰基还原为羟基,将羧基还原为醛基,但是与碳碳双键、碳碳三键都不发生 反应。
\ce{CH3CHO ->[NaBH4] CH3CH2OH}
四氢化锂铝 \ce{LiAlH4} 是非常强的还原剂,其给氢能力非常强,羧酸可以被其强制还原为醇。
\ce{R-COOH ->[LiAlH4][\text{无水}] R-CH2OH}
该反应通常在无水条件下进行。
硝基与氨基的转化及重氮化策略:硝基(\ce{-NO2})和氨基(\ce{-NH2})是一对可相互转化的官能团,在有机合成中扮演互补的角色。
- 硝基是强吸电子基,间位定位,可作为“临时指挥”引导取代反应,最终转化为氨基或其他官能团。
- 氨基是强给电子基,邻对位定位,既是重要的终产物,也是合成转换的枢纽。
硝基还原为氨基:
- 催化加氢:使用 \ce{H2} + \ce{Pd/C} 或 \ce{Pt}。条件温和,产率高,但如果分子内有碳碳双键或卤素(易脱卤),则需慎用。
- 金属还原:使用 \ce{Fe} 粉或 \ce{Sn} + \ce{HCl},是最经典的方法,但产生大量废物。
- 硫化物还原:使用 \ce{Na2S} 或 \ce{(NH4)2S},温和且可选择性还原多硝基化合物中的一个硝基。
重氮化反应:芳香伯胺与 \ce{NaNO2} + \ce{HCl} 在 0 \sim 5\pu{^oC} 冰水浴中反应生成重氮盐。重氮盐是一个“超级离去基团”(放出 \ce{N2}),可被多种亲核试剂取代:
- Sandmeyer 反应:加 \ce{CuCl}、\ce{CuBr} 或 \ce{CuCN},分别引入 \ce{-Cl}、\ce{-Br}、\ce{-CN}。
- Schiemann 反应:加 \ce{HBF4} 后加热分解,引入 \ce{-F}(芳氟化合物极难通过直接氟化合成)。
- 水解反应:加水加热,转化为酚 \ce{-OH}。
- 碘化反应:加 \ce{KI}(无需铜催化),引入 \ce{-I}。
- 脱氨基反应:加 \ce{H3PO2} 或乙醇,重氮基被 \ce{H} 取代。
- 偶合反应:与酚或芳香胺反应,生成偶氮化合物 \ce{-N=N-}(染料合成)。
硝基-氨基转化的关键合成策略:
定位效应的开关:硝基指引新基团进入间位,氨基指引新基团进入邻 / 对位。例如:
- 合成间溴苯胺:苯 \xrightarrow{\text{硝化}} 硝基苯 \xrightarrow{\ce{Br2/FeBr3}} 间溴硝基苯 \xrightarrow{\text{还原}} 间溴苯胺。
- 合成对溴苯胺:苯 \xrightarrow{\text{硝化}} 硝基苯 \xrightarrow{\text{还原}} 苯胺 \xrightarrow{\text{乙酰化保护}} 乙酰苯胺 \xrightarrow{\ce{Br2}} 对溴乙酰苯胺 \xrightarrow{\text{水解}} 对溴苯胺。
氨基作为占位基团:利用氨基的定位效应将目标基团“拉”到特定位置,再通过重氮化和脱氨基将氨基“抹除”。例如合成 \ce{1,3,5-} 三溴苯:苯胺加溴水直接生成 \ce{2,4,6-} 三溴苯胺,再重氮化后用 \ce{H3PO2} 去除重氮基。在此过程中,氨基完美扮演了“活化、占位、定位、事后消失”的工具人角色。
氨基的保护:游离氨基太活泼,直接溴化会导致三溴代,硝化等氧化条件下会被破坏。策略是通过酰化反应(如加乙酸酐)将 \ce{-NH2} 变成酰胺 \ce{-NHCOCH3},既降低活化能力又防止被氧化,待目标反应完成后再水解恢复。
常见试剂总结
[TODO]
“反应条件”,“反应类型” “氢氧化钠、水溶液、加热”,“卤代烃、酯酰胺的水解” “氢氧化钠、醇溶液、加热”,“卤代烃的消去” “浓硝酸、浓硫酸、加热”,“苯环上的硝化反应” “浓硫酸、加热”,“醇的消去反应或酯化反应及酯的水解反应” “氯气、液态溴、铁”,“苯环上的取代反应” “氧气、铜、加热”,“醇羟基的催化氧化” “银氨溶液、新制氢氧化铜”,“醛基的催化氧化” “氢气、镍催化”,“碳碳双键三键、醛酮、苯环与氢气的加成” “光照”,“烷烃的卤代反应”
使酸性高锰酸钾溶液褪色的:
分子中含有碳碳双键、碳碳三键的。
醇、酚、醛、酮、少数羧酸(草酸、甲酸等)。
苯的同系物,且与苯环直接相连的碳原子上有氢原子。
与羟基直接相连的碳原子上有氢原子的醇类物质。
具有还原性的无机还原剂。
使溴水褪色的:
分子中含有碳碳双键、碳碳三键的。
含有醛基的、所连碳原子的邻对位上有氢原子的酚类物质。
使溴水褪色几乎等同于使溴的四氯化碳溶液褪色,但醛基不能使溴的四氯化碳溶液褪色,因为醛基只有在水存在时,才得以被氧化成羧基。
特殊的,遇浓溴水产生白色沉淀,或者遇 \ce{FeCl3} 溶液显紫色的为酚羟基。
其他经典有机推断:
与氢气发生加成反应:碳碳双键、碳碳三键、醛基、酮羰基、苯。
遇三氯化铁溶液发生显色反应,或加入饱和溴水出现白色沉淀:酚羟基。
既能发生银镜反应,又能发生水解反应:\ce{-O-CHO}。
加入茚三酮溶液并加热,溶液显紫蓝色:氨基酸、蛋白质。
遇浓硝酸变黄:含有苯环结构。
按照产物推断:
若醇能被氧化为:
醛、羧酸:含有 \ce{-CH2OH} 结构。
酮:含有 \ce{>CHOH} 结构。
由反应确定官能团位置:
由消去反应的产物可确定 \ce{-OH} 或 \ce{C-X} 的大致位置。
由加氢或加溴后的碳骨架结构可确定 $\ce\ $ 或 \ce{C#C} 的位置。
由有机化合物发生酯化反应能生成环酯或高聚酯,可确定该有机化合物中含羟基和羧基,并根据酯环的大小,确定相对位置。
由取代产物的种类可确定碳骨架结构。
有机物溶解性:
亲水基越多越易溶于水,憎水剂越多、越大越难溶于水。
亲水基:羟基(\ce{-OH}),醛基(\ce{-CHO}),羧基(\ce{-COOH}),氨基(\ce{-NH2}),酚(\ce{C6H5OH})。
憎水基:烃基(\ce{-C_nH_{2n+1}},\ce{-CH=CH2},\ce{-C6H5}),卤素(\ce{-X}),硝基(\ce{-NO2}),酯基(\ce{-COO-})。
例如:一般的,\ce{C} 原子个数大于 5 的醇不溶于水,小于等于 4 则可能溶于水。
有机物之间:含有相同官能团的物质一般可以互溶。
有机溶剂(\ce{C6H6},\ce{C2H4},\ce{CCl4},\ce{C2H5OH})可以溶解绝大多数有机物。
定量关系总结
烃和卤素单质的取代:
取代 \pu{1mol} 氢原子,消耗 \pu{1mol} 卤素单质、
碳碳双键的加成:
与 \ce{H2、Br2、HCl、H2O} 等加成时按物质的量之比为 1:1 反应、
含 \pu{1mol} 醇羟基的有机物:
- 与 \ce{Na} 反应:生成 \pu{0.5mol}\ce{H2}。
含 \pu{1mol} 酚羟基的有机物:
与 \ce{Na} 反应:生成 \pu{0.5mol}\ce{H2}。
与 \ce{NaOH} 反应:生成 \pu{1mol}\ce{H2O}。
与 \ce{Na2CO3} 溶液产生 \pu{1mol}\ce{NaHCO3}。
含 \pu{1mol} 羧基的有机物:
与 \ce{Na} 反应:生成 \pu{0.5mol}\ce{H2}。
与 \ce{NaOH} 反应:生成 \pu{1mol}\ce{H2O}。
与 \ce{NaHCO3} 溶液产生 \pu{1mol}\ce{CO2}。
含 \pu{1mol} 醛基的有机物:
与 \pu{2mol}\ce{[Ag(NH3)2]OH} 反应,生成 \pu{2mol}\ce{Ag}。
与 \pu{2mol}\ce{Cu(OH)2} 反应,生成 \pu{1mol}\ce{Cu2O}。
注意:\pu{1mol} 甲醛含 \pu{2mol}\ce{-CHO}。
有机物与 \ce{NaOH}(水解)的定量关系:
\pu{1mol} 酯键 / 酰胺键与 \pu{1mol}\ce{NaOH} 反应。
\pu{1mol} 碳卤键与 \pu{1mol}\ce{NaOH} 反应。
注意:酚酯等有多个符合条件的物质,既有水解又有中和。
注意:若卤素原子取代在苯环上,碳卤键水解后能还能进行酚的中和,与 \pu{2mol}\ce{NaOH} 反应。
物质转化过程中相对分子质量的变化:
- 醇、醛、酸的连续氧化:M \to (M-2) \to (M+14)。
互变异构概述
羰基-烯醇互变异构:
互变的核心是 \alpha-氢(羰基旁边碳上的氢)转移到氧上,同时双键从 \ce{C=O} 变成 \ce{C=C}。
过程需要催化剂加速,比如酸或碱。没有催化剂,也能发生,但很慢。
绝大多数情况下,酮式更加稳定,除非烯醇式分子内可以形成氢键或苯环,例如苯酚。
环-链互变异构:
环-链互变异构是整个分子在开环(链状)和闭环(环状)结构之间的平衡。
通常发生在同一个分子内含有两个可以缩合的官能团,且两者位置合适(通常能形成较为稳定的五元环或六元环)。
典型例子:在水溶液中,葡萄糖绝大部分不是以我们常画的链状结构存在,而是以环状的半缩醛形式存在。
理解环-链互变异构的关键在于掌握半缩醛与缩醛的概念。
半缩醛的通式为 \ce{R2C(OH)OR'},即同一个碳原子上同时连接 -\ce{OH} 和 -\ce{OR'}。它通常是由醛或酮与一分子醇在酸催化下发生亲核加成得到的中间体,多数为不稳定的平衡体系。
缩醛的通式为 \ce{R2C(OR')2},即同一个碳原子上连接两个 -\ce{OR'} 基团(同碳二醚),可视为半缩醛再与一分子醇脱水缩合的产物。
关键反应流程:羰基 + 醇 \xrightarrow{\text{酸催化}} 半缩醛 \xrightarrow{\text{酸催化}} 缩醛 + \ce{H2O}。缩醛化是可逆平衡反应,推动平衡需使用过量醇并移走水。
稳定性规律:缩醛在中性和碱性条件下很稳定(像醚一样),但在水相酸中会水解回原来的羰基化合物和醇。因此缩醛常作为羰基保护基使用。
糖化学中的重要性:单糖(如葡萄糖)在水溶液中主要以环状半缩醛形式存在(五元或六元环),那个特殊的碳原子称为异头碳。两个糖通过异头位的半缩醛羟基脱水形成糖苷键(本质就是缩醛结构),如蔗糖中葡萄糖部分为缩醛、果糖部分为缩酮。
有机化学中有三大主要的电子效应:
诱导效应(I 效应):由于成键原子电负性差异,电子云沿 \sigma 键偏移。短程效应,通常经过 3 个碳原子后可忽略。分为 -\ce{I}(吸电子,如 -\ce{NO2}、-\ce{CN}、卤素)和 +\ce{I}(给电子,如烷基)。
共轭效应(C 效应):在共轭体系中,\pi 电子或 p 电子的离域导致电子云重新分布。长程效应,只要共轭体系不中断就不衰减。分为 -\ce{C}(吸电子,如 -\ce{NO2}、-\ce{CHO})和 +\ce{C}(给电子,如 -\ce{NH2}、-\ce{OH}、卤素的孤对电子)。
超共轭效应(H 效应):\sigma 键(通常是 \ce{C-H})的电子与相邻 \pi 轨道或空 p 轨道的部分重叠。效应强弱取决于 \alpha-\ce{H} 的数量,\alpha-\ce{H} 越多超共轭越强。碳正离子稳定性 3° > 2° > 1° 即源于此。
强度顺序:共轭效应(C)> 诱导效应(I)> 超共轭效应(H)。
经典案例:
-\ce{OH} 和 -\ce{NH2}:-\ce{I} 吸电子,但 +\ce{C} 给电子更强(+\ce{C} > -\ce{I}),总体为强给电子基团,使苯环活化。
卤素(-\ce{F}、-\ce{Cl}、-\ce{Br}、-\ce{I}):-\ce{I} 强吸电子,+\ce{C} 弱给电子(-\ce{I} > +\ce{C}),总体为吸电子基团(使苯环致钝),但因 +\ce{C} 效应稳定邻对位中间体,仍为邻对位定位基。卤素(-\ce{Cl}、-\ce{Br} 等)是苯环定位效应中最具代表性的「矛盾体」:它使苯环钝化(-\ce{I} 效应吸走电子,降低亲电取代活性),却同时将新取代基引导到邻位和对位(+\ce{C} 效应的孤对电子在亲电进攻时可稳定邻对位中间体)。这打破了「活化 = 邻对位,钝化 = 间位」的简单对应,说明定位效应和活化/钝化是两个独立维度。
另一个常见反例是苯胺在强酸中的硝化。\ce{-NH2} 本是强邻对位定位基,但在浓硫酸 / 浓硝酸的强酸环境中,\ce{-NH2} 被质子化为 \ce{-NH3+},后者凭借正电荷成为强吸电子基,使苯环钝化并转为间位定位,导致产物以间硝基苯胺为主。
类似地,苦味酸(2,4,6-三硝基苯酚)虽然只是一个酚(连 \ce{-OH}),但其酸性竟强于乙酸。这是因为三个强吸电子硝基将 \ce{-OH} 脱质子后的负电荷高度分散,阴离子极稳定,导致它极容易给出 \ce{H+}。
在普通的脂肪族亲核取代反应中,离去能力顺序为 \ce{I > Br > Cl \gg F},因为 \ce{C-F} 键极强,氟是最差的离去基团。但在**芳香族亲核取代反应(\mathrm{S_NAr})**中,当苯环上带有强吸电子基时,含氟底物的反应速度反而是最快的,\ce{Ar-F \gg Ar-Cl > Ar-Br > Ar-I}。
这是因为 \mathrm{S_NAr} 的决速步不是断裂碳卤键,而是亲核试剂进攻苯环形成梅森海默络合物。氟的电负性最强,将苯环电子云大幅拉向自身,使被进攻碳的正电性最强,最有利于亲核试剂的攻击。因此在这一机理下,氟的「离去能力」由它促进亲核进攻的能力决定,而非碳卤键的强弱。
在多步有机合成中,官能团保护是一个核心策略:当分子中存在多个反应活性中心时,为防止试剂与不该反应的官能团发生副反应,需要将其暂时保护,待目标反应完成后再脱保护。
好保护基的四条标准:易于引入、足够稳定(耐受后续反应条件)、易于脱除(条件专一)、副产物易分离。
常见保护策略:
醇羟基:硅醚类(TBS 用氟离子脱除,TBDPS 更稳定)、苄醚(催化氢解脱除)。
氨基:Boc(酸脱除,对碱稳定)、Cbz(催化氢解脱除)、Fmoc(碱脱除,对酸稳定)。
羰基:缩醛 / 缩酮(对碱和亲核试剂稳定,酸性水解脱除)。
羧基:甲酯 / 叔丁酯 / 苄酯(水解或氢解脱除)。
正交保护:在同一分子中使用脱除机制完全不同的保护基,可以按特定顺序依次脱除而不互相干扰。例如 Boc(酸脱)与 Fmoc(碱脱)在多肽合成中的经典组合。
在物质推断题中,不同物质的相对分子质量(M_r)可能相等,需结合化学性质区分。
无机物常见等 M_r 对:
| M_r | 物质 A | 物质 B | 区分要点 |
|---|---|---|---|
| 28 | \ce{CO} | \ce{N2} | \ce{CO} 可燃、有还原性;\ce{N2} 惰性 |
| 44 | \ce{CO2} | \ce{N2O} | \ce{CO2} 使石灰水浑浊;\ce{N2O} 支持燃烧 |
| 78 | \ce{Na2S} | \ce{Na2O2} | \ce{Na2S} 放 \ce{H2S};\ce{Na2O2} 放 \ce{O2} |
有机物常见等 M_r 对:
| M_r | 物质 A | 物质 B | 区分要点 |
|---|---|---|---|
| 44 | \ce{C3H8}(丙烷) | \ce{CH3CHO}(乙醛) | 乙醛可银镜反应 |
| 46 | \ce{C2H5OH}(乙醇) | \ce{CH3OCH3}(二甲醚) | 乙醇与 \ce{Na} 放 \ce{H2} |
| 58 | \ce{CH3COCH3}(丙酮) | \ce{CH3CH2CHO}(丙醛) | 丙醛可银镜反应 |
N- 前缀表示取代基连接在氮原子上,而非碳链上。这在酰胺和胺的命名中非常常见。
例如 N-甲基甲酰胺 \ce{HCONHCH3},即甲酰胺 \ce{HCONH2} 中氮上的一个氢被甲基取代。
常见类型:
二级酰胺 \ce{RCONHR'}:命名为 N-某基某酰胺。
三级酰胺 \ce{RCONR'2}:命名为 N,N-二某基某酰胺。
仲胺 \ce{RNHR'}:命名为 N-某基某胺。
官能团优先顺序决定母体名称:羧酸 > 酯 > 酰胺 > 醛 > 酮 > 醇 > 胺 > 烯烃 / 炔烃。优先级最高的官能团决定词尾,其余为取代基。
Diels-Alder 反应(DA 反应)是有机化学中最重要的成环反应之一,能够一步同时构建两个 C–C 键和一个六元环。
基本定义:反应本质是 [4+2] 环加成,共轭双烯体提供 4 个 \pi 电子,亲双烯体(通常为烯烃或炔烃)提供 2 个 \pi 电子,经协同过渡态生成环己烯衍生物。
反应物要求:
双烯体必须能采取 s-cis 构象(被锁死为 s-trans 的双烯无法反应)。
亲双烯体通常带有吸电子基团(-\ce{C=O}、-\ce{CN}、-\ce{NO2}),使其缺电子,与富电子双烯匹配。
三大核心规则:
立体专一性:亲双烯体上取代基的顺反关系在产物中保持不变。
内型规则(Endo Rule):当形成双环体系时,亲双烯体上的吸电子基团倾向于折叠在双烯 \pi 体系正下方,生成动力学优势的内型(Endo)产物,原因是过渡态中存在次级轨道相互作用。
区域选择性:不对称底物倾向于生成「邻位」(1,2-取代)或「对位」(1,4-取代)产物,极少生成间位产物。
重要延伸:
逆 DA 反应(Retro-DA):高温下六元环可裂解回双烯体和亲双烯体,用于保护基技术和自修复高分子材料。
逆电子需求 DA(IEDDA):双烯体缺电子、亲双烯体富电子时发生。四嗪与反式环辛烯的 IEDDA 反应是已知最快的生物正交反应之一,广泛应用于活体成像和药物靶向追踪。
羰基化合物与 HCN 加成再水解是经典的「一碳增长」路线,最终生成 \alpha-羟基酸。
第一步:亲核加成生成氰醇
醛或酮在弱碱性条件下受 \ce{CN^-} 攻击,生成 \alpha-羟基腈(氰醇)。现代合成中常用三甲基硅基氰(TMSCN)代替剧毒的 HCN 气体,安全且产率高。
第二步:酸性水解机理
氰基(-\ce{C#N})经酸催化水解转化为羧基(-\ce{COOH}),过程分为两个阶段:
氰基转化为酰胺:\ce{H^+} 首先质子化氰基氮原子,增强碳的亲电性;\ce{H2O} 亲核攻击后经互变异构生成伯酰胺(-\ce{CONH2})。
酰胺转化为羧酸:酰胺羰基氧被质子化,水分子进攻形成四面体中间体;-\ce{NH2} 被质子化为 -\ce{NH3^+}(优良离去基团),离去后生成羧酸,氮以铵盐形式离开。
经典应用——Strecker 氨基酸合成:
将醛与 \ce{HCN} 和 \ce{NH3} 混合,先生成亚胺再加成 HCN,得到 \alpha-氨基腈,水解后即得 \alpha-氨基酸。这是工业上合成氨基酸的经典方法之一。
羟醛缩合是构建 C–C 键最核心的反应之一,通过羰基化合物 \alpha-位的活化实现碳链延长与官能团转化。
核心前提:至少有一方反应物存在 \alpha-H(羰基相邻碳上的氢)。\alpha-H 因羰基的吸电子效应和烯醇负离子的共振稳定而呈微酸性。
反应分两步进行:
羟醛加成:碱夺取 \alpha-H 生成烯醇负离子,亲核攻击另一分子羰基碳,形成 \beta-羟基醛(或酮)。
脱水缩合:\beta-羟基产物在加热或酸碱条件下脱水,生成 \alpha,\beta-不饱和羰基化合物。脱水的热力学驱动力是共轭体系(\ce{C=C-C=O})的形成。
交叉羟醛缩合策略:
- 若两反应物都有 \alpha-H,产物混合严重。实用策略是使用无 \alpha-H 的羰基化合物(如苯甲醛、甲醛)作为亲电受体,保证单一产物。经典案例如 Claisen-Schmidt 缩合(苯甲醛与丙酮生成苄叉丙酮)。
分子内羟醛缩合:
- 同一分子内含有两个羰基和合适的 \alpha-H 时,可发生分子内关环。产物受张力控制,几乎总是生成五元环或六元环。