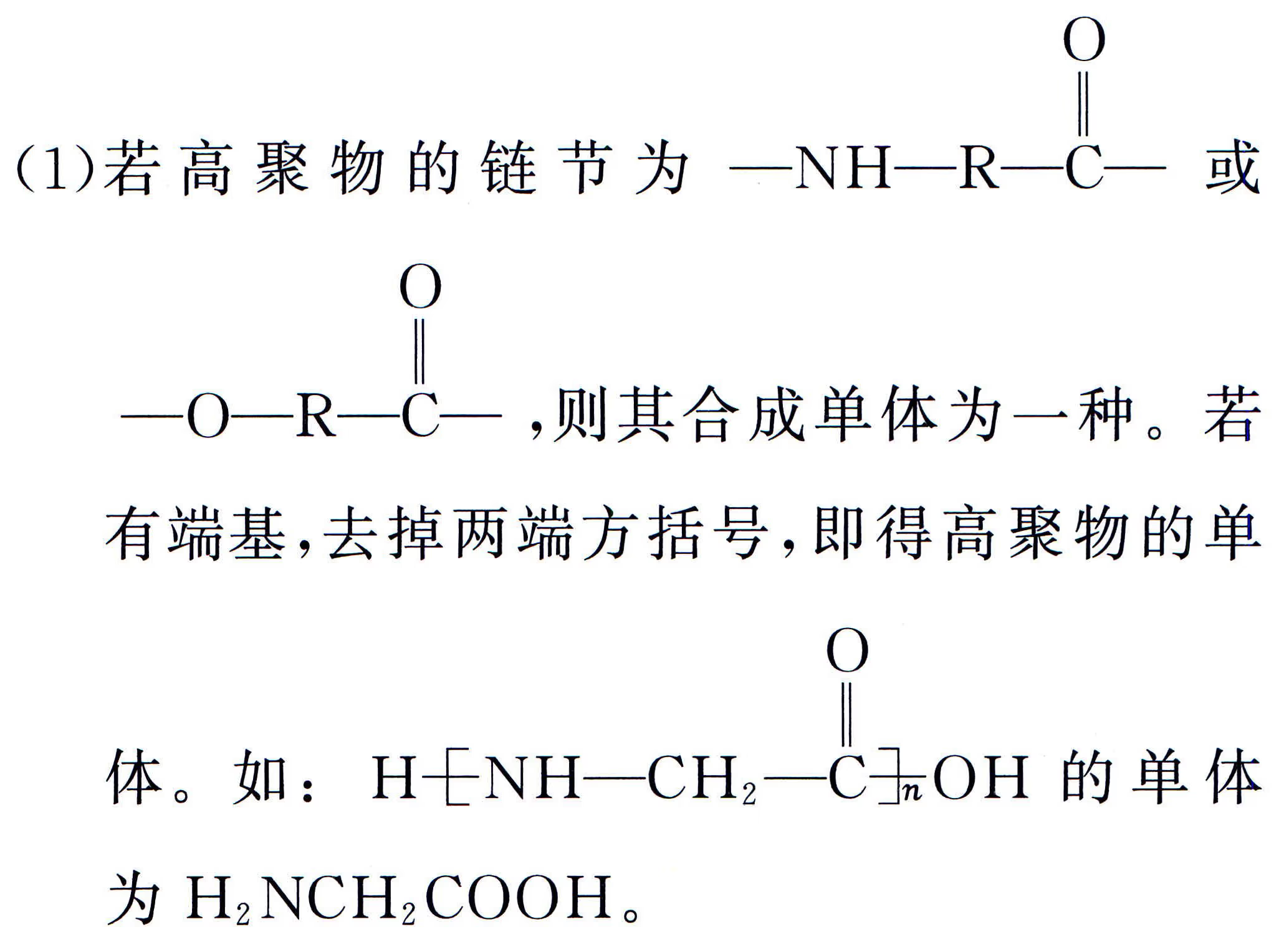
有机物与工业
高分子概述
有机高分子
高分子化合物,简称高分子,相对分子质量通常在几万到几十万,甚至达到几千万。高分子虽然相对分子质量很大,但结构往往并不复杂。
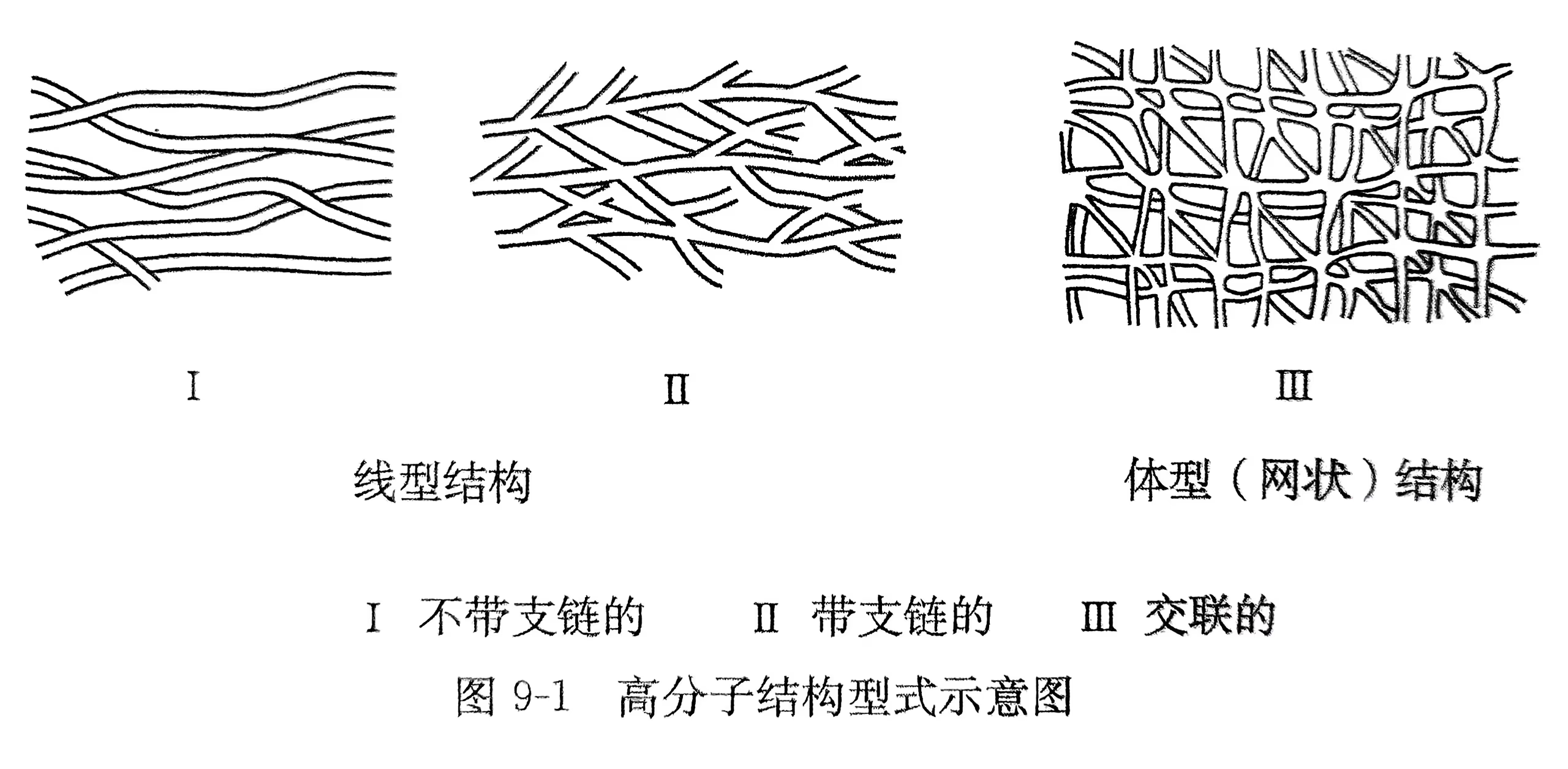
高分子的性质:
在溶剂中的溶解。
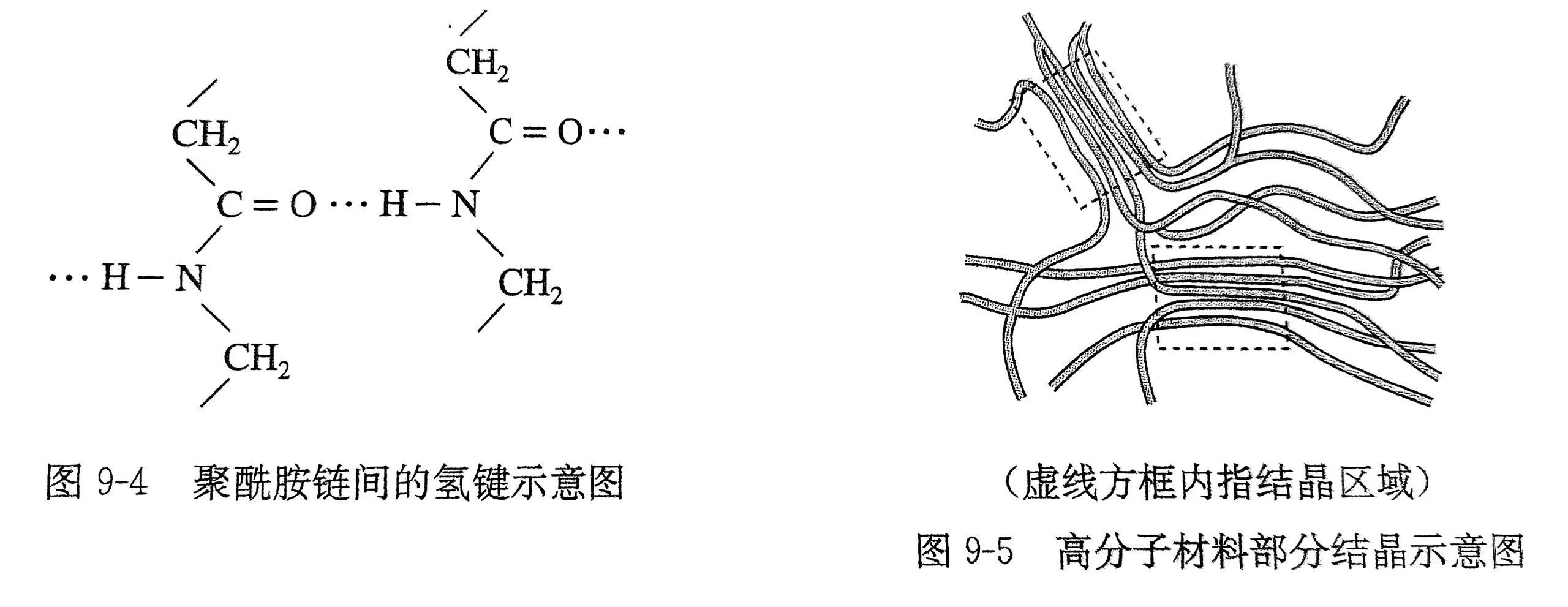
高分子溶解在有机溶剂中,溶剂分子先深入缠在一起的线性高分子之间,使高分子材料膨胀,然后把高分子包围起来,使其分离开来,能够自由移动,在有机溶剂中形成胶体粒子,成为溶胶。
不同温度下的性能。
热塑性:高分子受热到一定温度范围,开始软化,直到熔化成流动的液体,不像固态小分子物质那样有确定的熔点。加热软化后,可以加工成各种形态。
热固性:网状结构中的高分子链已有化学键相互交联,限制了其移动。当温度更高,体系高分子的化学键出现断裂,高分子的结构就被破坏。一经加工成形,就不会受热融化,如酚醛塑料。
强度和电绝缘性。
高分子材料的密度比金属小,但强度可能比较大。
大部分有机高分子材料是绝缘的,但也有导电的,如单双键交替的碳链。
高分子材料
三大天然有机高分子:淀粉、纤维素、蛋白质。
三大合成材料:塑料、合成纤维、合成橡胶。
塑料:
例如:合成树脂,如聚乙烯、聚氯乙烯、聚四氟乙烯。树脂的含义是未加工的处理物。
聚乙烯制成的塑料是热塑性塑料,而酚醛树脂等只能一次成形,是热固性塑料。具有网状结构的高分子受热都不能软化或熔融,也不溶于 任何溶剂。
聚四氟乙烯:塑料王,除熔融的碱金属和液氟外,几乎不受任何化学试剂的腐蚀,甚至放在王水中煮沸,其性能也不会发生改变。
不能用含增塑剂的聚氯乙烯薄膜等做食品包装材料。
超高相对分子质量(大于 100 万)的聚乙烯可用作防弹头盔和防弹衣的材料。
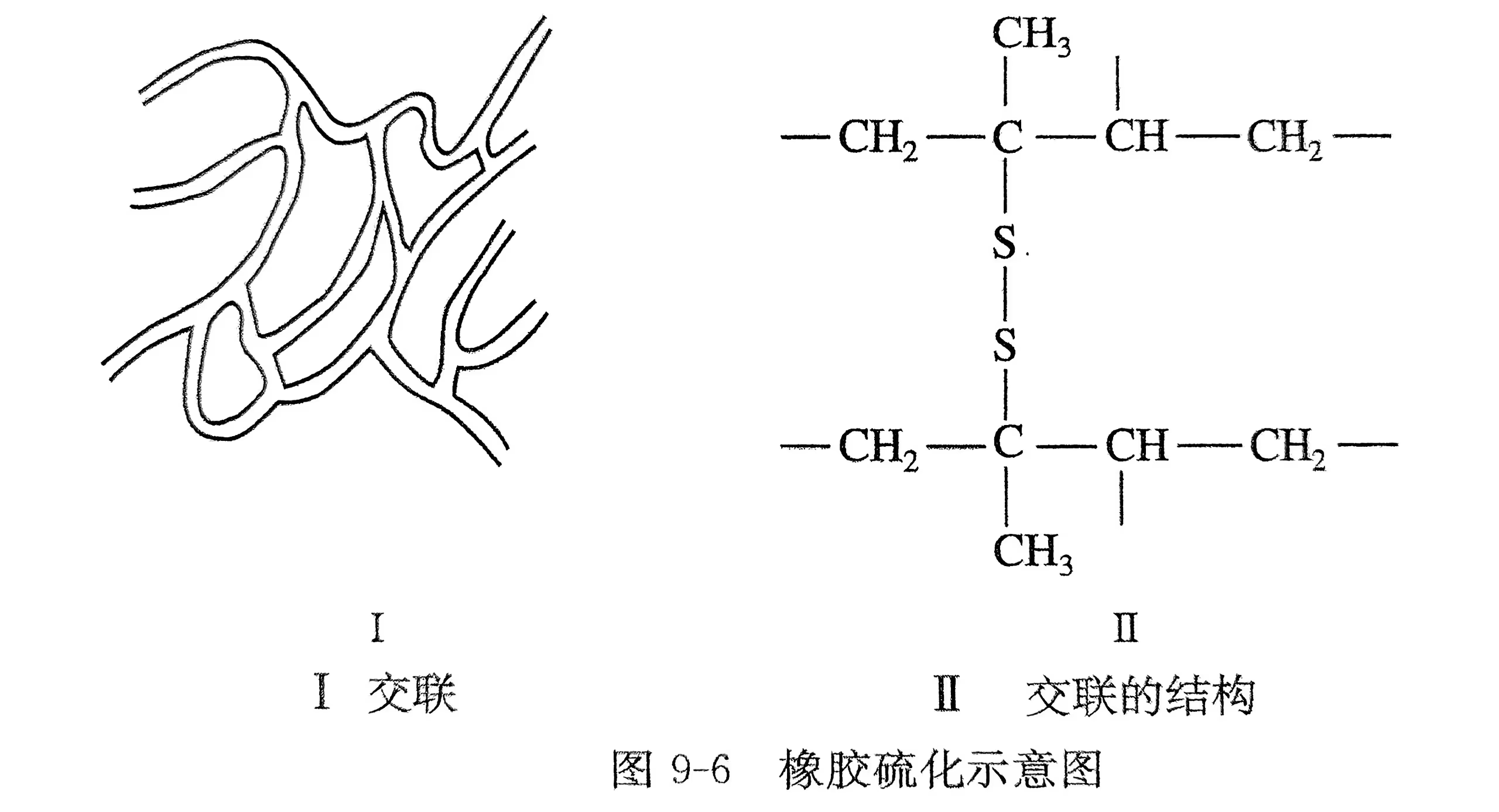
橡胶:
橡胶是一种具有高弹性的高分子材料。
天然橡胶的成分主要是顺式聚异戊二烯。
合成橡胶主要有:顺丁橡胶、丁苯橡胶、丁腈橡胶、乙丙橡胶、硅橡胶等。
合成纤维:
纤维,分为天然纤维(蚕丝、羊毛、棉花、天然纤维纺纱)和化学纤维,其中化学纤维又分为人造纤维(再生纤维)和合成纤维。
棉花、羊毛、蚕丝和麻等是大自然赋予人们的天然纤维。
以木材、秸秆等农副产品为原料,经加工处理可以得到再生纤维。
以石油、天然气、煤、农副产品为原料,将其转化为单体,再经过聚合反应得到的是合成纤维。
功能高分子材料:
高吸水性材料。
高分子分离膜。
医用高分子材料。
聚合反应
加聚反应
聚合反应:
乙烯可以和自己聚合,生成环丁烷、环己烷等;当聚合的长度足够长,就难以形成环,即聚乙烯。
在适当的温度、压强和催化剂存在的条件下,乙烯分子中碳碳双键的一个键断裂,分子间通过碳原子相互结合形成很长的碳链,生成相对分子质量很大的聚合物——聚乙烯。
\ce{n CH2=CH2 ->[催化剂] \underset{聚乙烯}{\poly{CH2-CH2}}}
像这样,由相对分子质量小的化合物分子相互结合成相对分子质量大的聚合物的反应叫做聚合反应。乙烯的聚合反应同时也是加成反应,这样的反应又被称为加成聚合反应,简称加聚反应,通常没有副产物生成。
- 单体:能合成高分子化合物的小分子物质,如乙烯(\ce{CH2=CH2})。
- 链节:聚合物中重复的结构单元,如聚乙烯中的“-\ce{CH2-CH2}-”。
- 聚合度:链节的数目 n。
- 聚合物(高分子化合物):反应的生成物,如聚乙烯(\ce{-[CH2-CH2]_n-})。
经典的加聚反应:
炔烃的加聚:
\ce{n CH2\bond{3}CH2 ->[催化剂] \underset{聚乙炔}{\poly{CH2=CH2}}}
二烯烃加聚:
\ce{n CH2=CH-CH=CH2 ->[催化剂] \poly{CH2-CH=CH-CH2}}
烯烃共聚:
\ce{n CH2=CH2 + n CH2=CH-CH3 ->[一定条件] \poly{CH2-CH2-CH(CH3)-CH2}}
缩聚反应
由有机化合物分子间脱去小分子获得高分子化合物的反应称为缩合聚合反应,简称缩聚反应。
- 缩聚反应生成聚合物的同时,还有小分子副产物(如 \ce{H2O} 等)生成。
- 缩聚反应的单体通常是具有两个或多个官能团(如 \ce{-OH},\ce{-COOH},\ce{-NH2} 等)的小分子。
- 所得聚合物链节的化学组成与单体的化学组成不同。
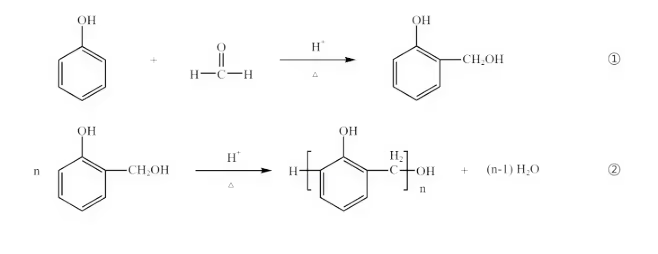
经典的缩聚反应:
酚醛树脂的合成:
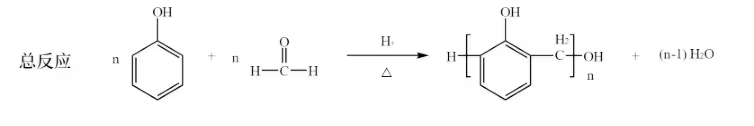
羟基酸缩聚:


醇酸缩聚:


氨基酸缩聚:
\ce{n H2N(CH2)5COOH <=>[一定条件] H\poly{NH(CH2)5CO}OH + $(n-1)$ H2O}
结构判断
加聚产物单体判断:要先判断高分子化合物是加聚产物还是缩聚产物。
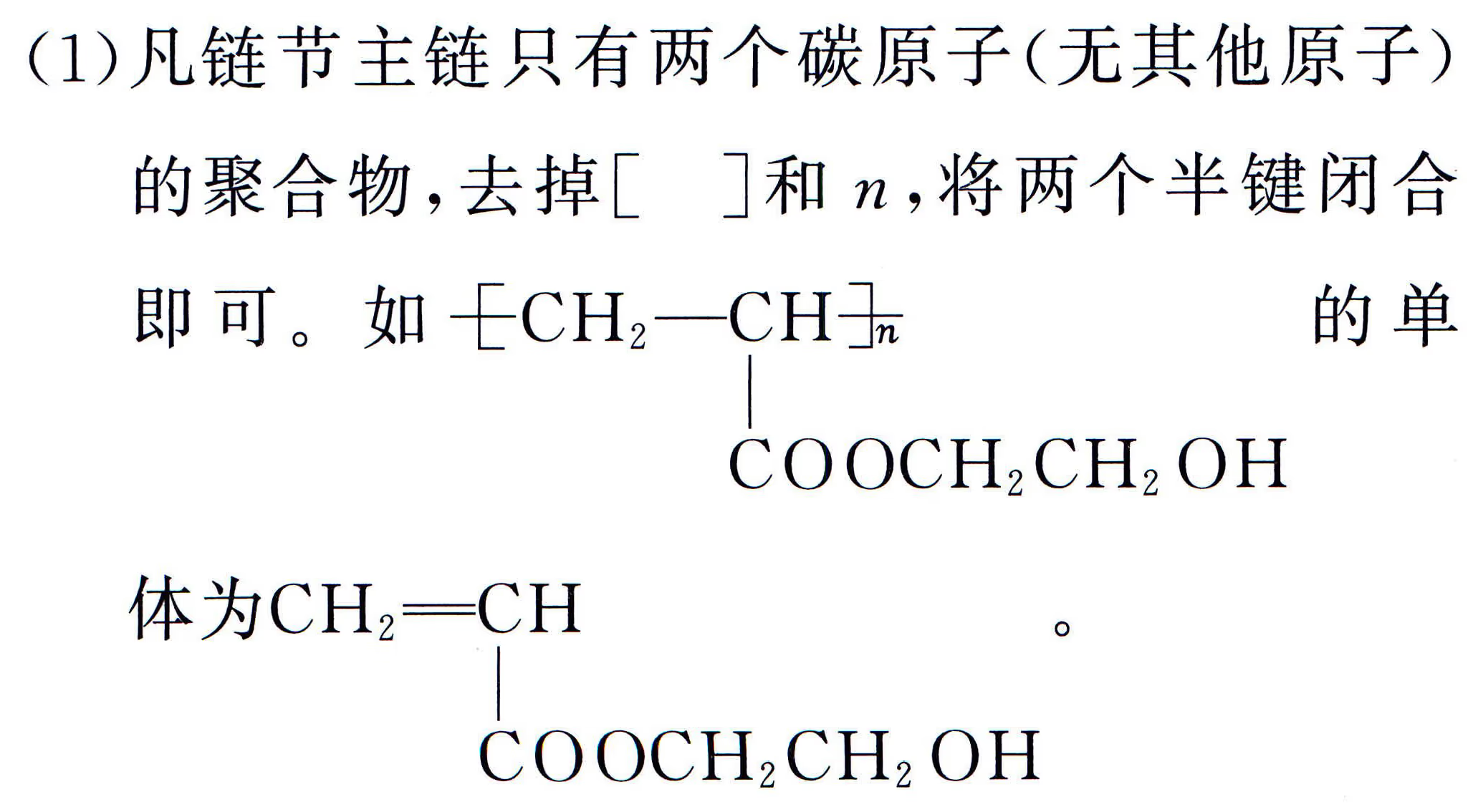

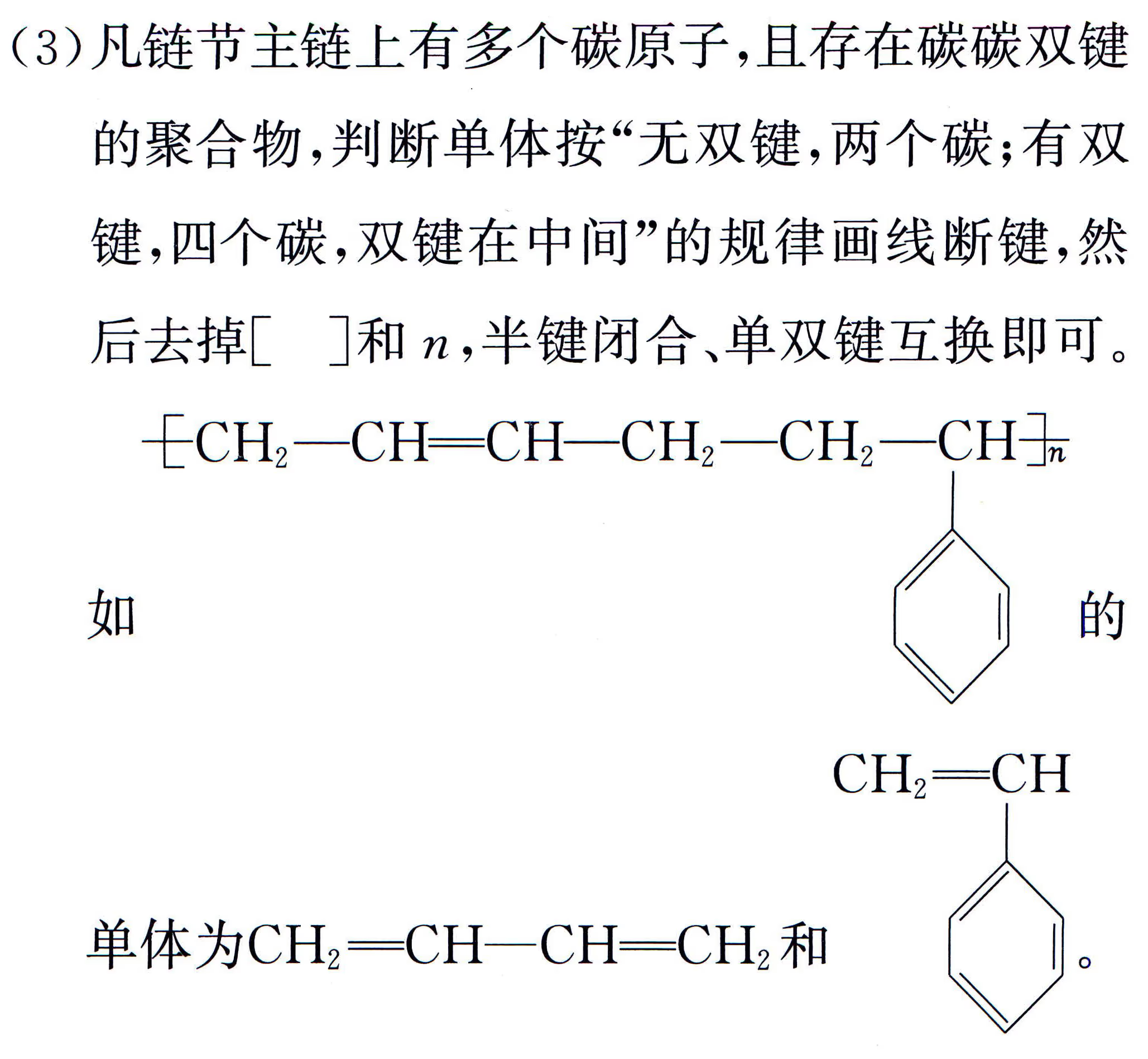
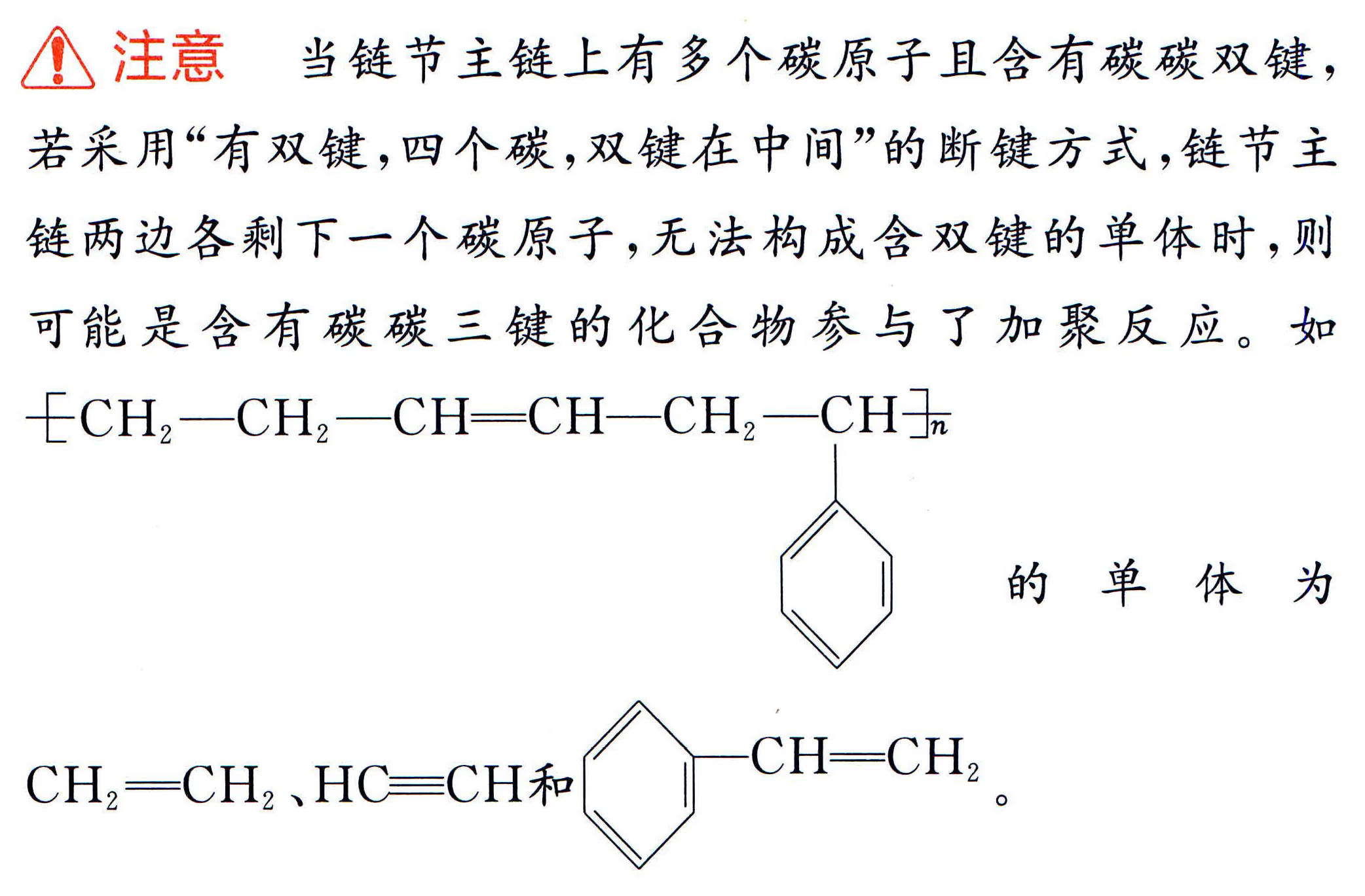
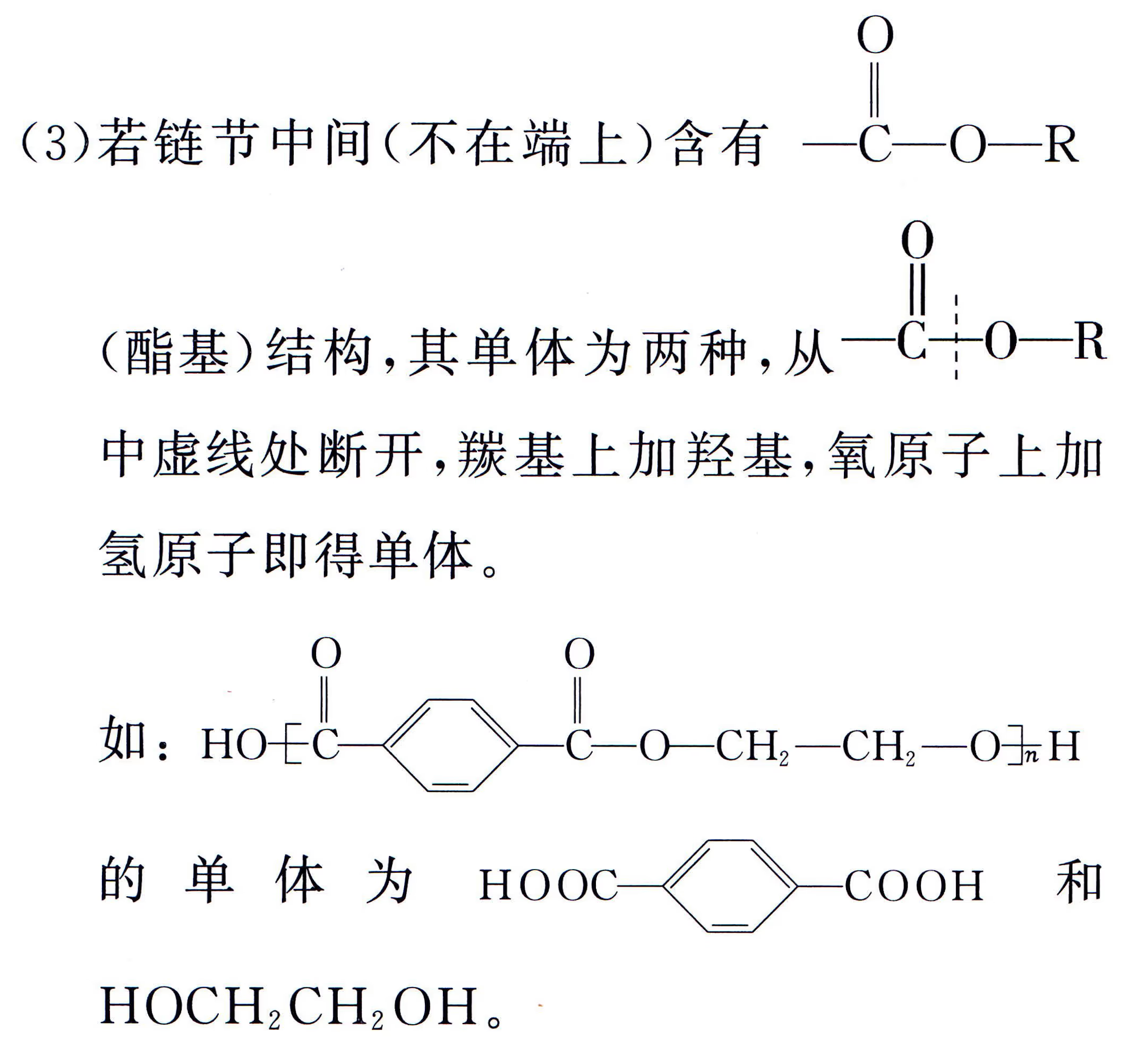
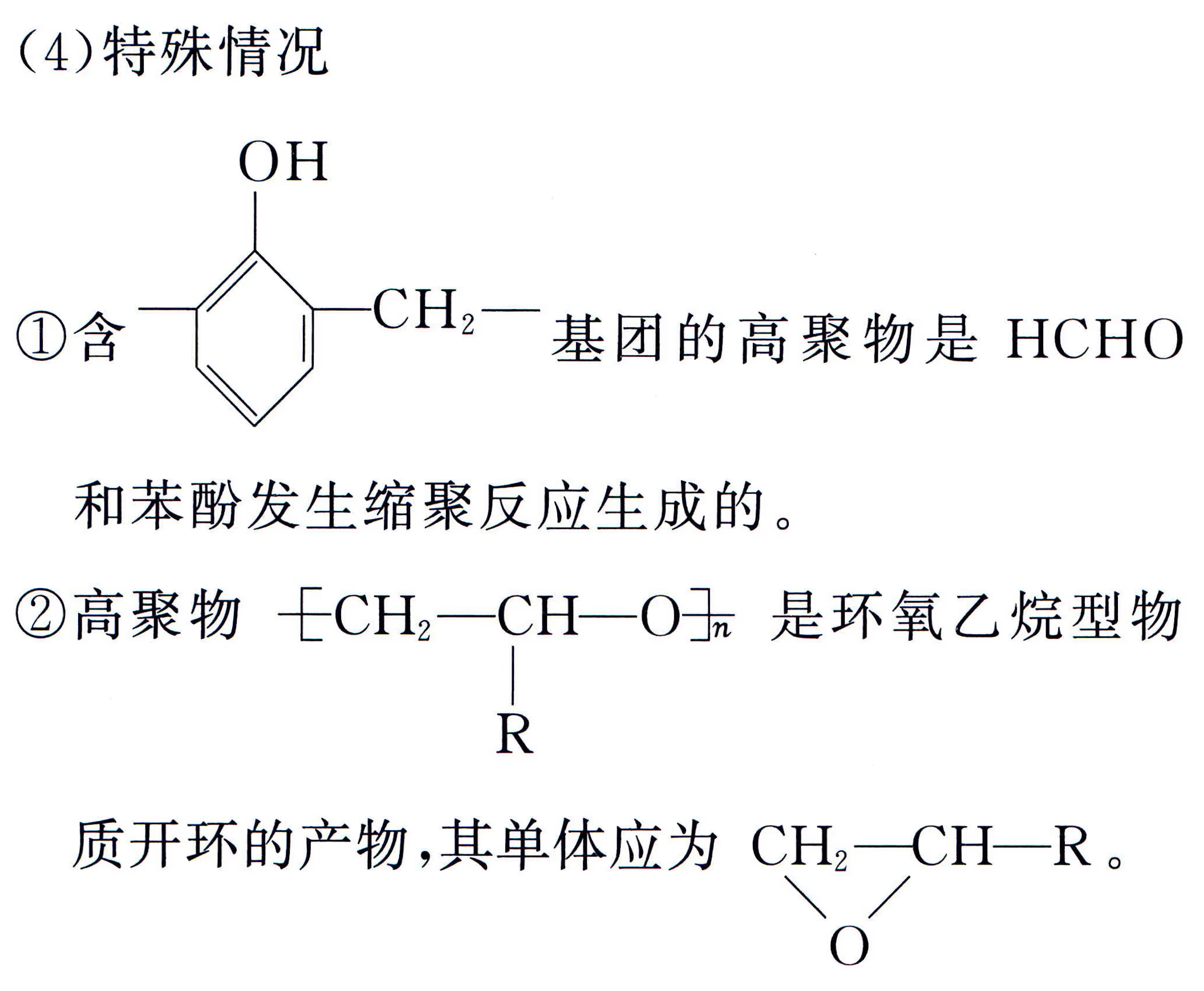
缩聚产物单体判断:中学里缩聚产物形成的方式有三种:聚酯式、聚酰胺式、酚醛树脂式,从形式上看都是脱水缩合。

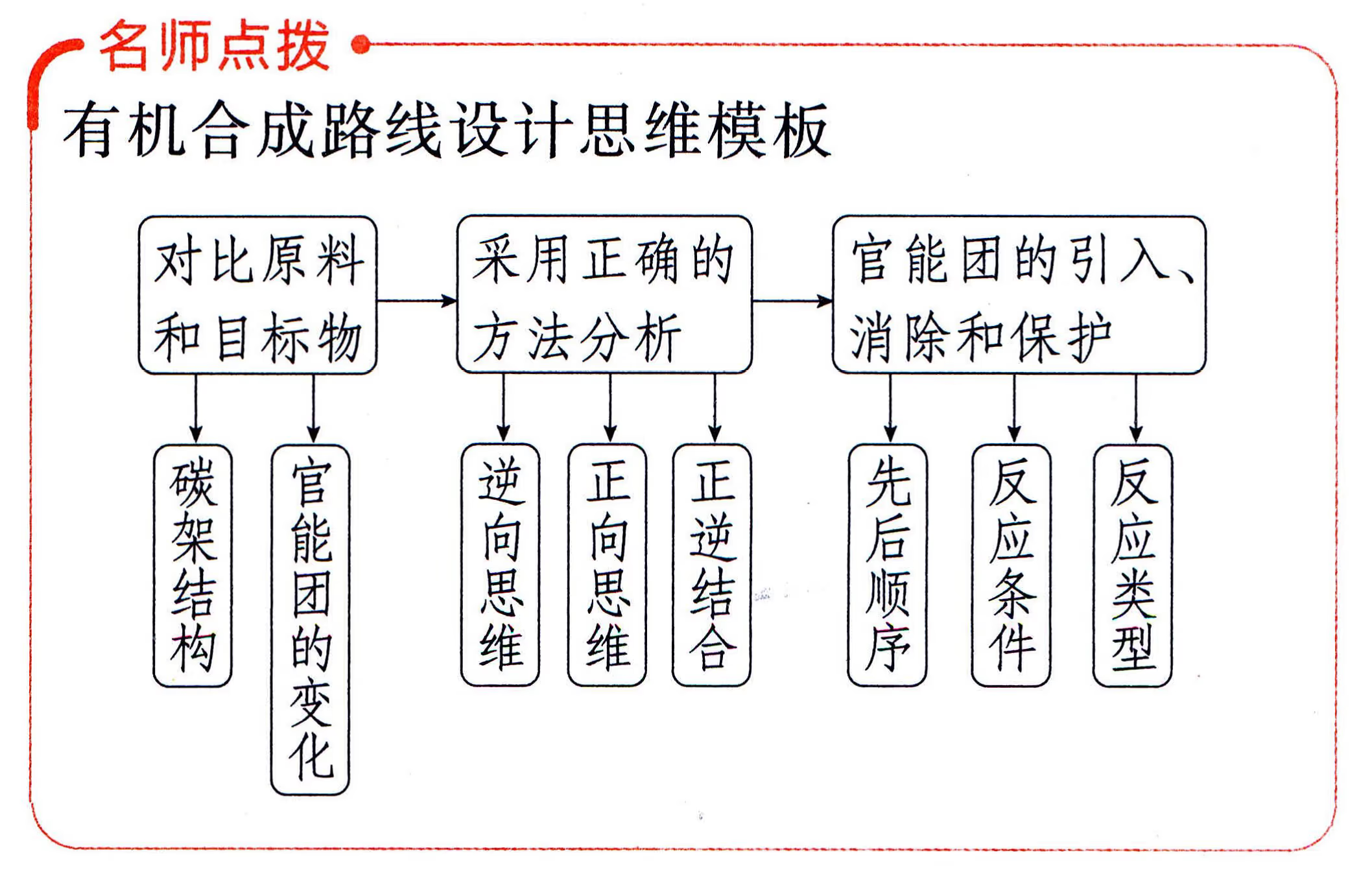
正合成分析法:采用正向思维方法,从已知原料入手,找出合成所需要的直接或间接的中间体,逐步推向待合成的有机物。其思维程序是:原料→中间体→产品。
逆合成分析法:采用逆向思维方法,从产品的组成、结构、性质入手,找出合成所需要的直接或间接的中间体,逐步推向原料。其思维程序是:产品→中间体→原料。
类比合成分析法:比较题目所给有机合成路线及已知信息反应,找出原料与合成物质的内在联系,确定中间产物,最后得到目标产物——产品。
从聚合物结构反推单体
有时候题目会给出聚合物的循环节,要求你反推基础单体。解决这类问题的关键是识别循环节中各结构片段的化学来源。
例 已知某树脂的循环节为:-\ce{CH2}-[\text{苯环 (邻位连链、2 位酚羟基)}]-\ce{CH2}-\ce{N}[\text{苯环 (3 位甲基)}]-,判断其基础单体。
分析:
两个 -\ce{CH2}-(亚甲基桥)来自甲醛(\ce{HCHO})。
含酚羟基的苯环片段来自苯酚(\ce{C6H5OH})。
氮上连接的间甲苯基来自间甲苯胺(3-甲基苯胺,\ce{CH3-C6H4-NH2})。
因此,基础合成单体为苯酚 + 甲醛 + 间甲苯胺。这三种原料先通过 Mannich 缩合生成苯并噁嗪单体,再经热开环聚合得到目标结构。
甲醛在缩聚中产生亚甲基的原因
在酚醛树脂、脲醛树脂等含甲醛的缩聚反应中,最终出现的**亚甲基(\ce{-CH2-})**本质上就是甲醛那一个碳留下来的结果。
甲醛在缩聚里通常不是一步直接变成桥,而是分两步:
- 羟甲基化:活泼位点先和甲醛反应,生成羟甲基化产物(\ce{-CH2OH})。
- 脱水桥连:羟甲基再和另一个活泼位点缩合,脱去一分子水,碳保留下来形成 \ce{-CH2-} 桥。
以苯酚为例:
\ce{PhOH + HCHO -> HO-Ph-CH2OH}
\ce{HO-Ph-CH2OH + PhOH -> HO-Ph-CH2-PhOH + H2O}
甲醛之所以比其他醛更容易形成这种桥,是因为:
- 位阻最小:甲醛没有烷基取代基,羰基碳更「裸露」,更容易被进攻。
- 亲电性强:其他醛(如乙醛)会给 \ce{-CH(CH3)-} 桥,反应慢、位阻大、产物规整性差。
- 最短桥:甲醛只给一个 \ce{C1} 桥,交联密度高、网络更紧密、耐热性更好。
注意区分:含苯环的缩聚不一定产生亚甲基桥。例如 PET(对苯二甲酸 + 乙二醇)产生酯键,芳纶产生酰胺键。真正容易形成亚甲基桥的条件是甲醛参与且苯环上有活化基团(\ce{-OH}、\ce{-NH2})。
酚醛树脂
酚醛树脂(Phenol-Formaldehyde Resin,简称 PF),俗称电木,是人类历史上第一种完全人工合成的高分子塑料。其合成基于苯酚与甲醛的缩聚反应,根据催化剂的不同,可分为碱催化体系(热固性树脂)和酸催化体系(热塑性树脂)。
碱催化体系(Resol / 甲阶树脂)
反应条件:甲醛过量(苯酚 : 甲醛摩尔比通常为 1 : 1.2 \sim 1.5),催化剂为碱(如 \ce{NaOH}、\ce{NH3.H2O})。
反应机理:
- 碱首先夺取苯酚上的质子,生成苯氧负离子(\ce{PhO-}),其亲核性远强于苯酚。
- 活化的邻 / 对位碳攻击甲醛的羰基碳,发生亲核加成反应,生成邻羟甲基苯酚或对羟甲基苯酚。由于甲醛过量,会进一步生成二羟甲基、甚至三羟甲基苯酚。
- 加热条件下,羟甲基之间脱水形成醚键(\ce{-CH2-O-CH2-}),或羟甲基与苯环上的活泼氢脱水形成亚甲基桥(\ce{-CH2-})。
固化:将甲阶树脂加热至 150 \sim \pu{200^oC},残留的羟甲基继续发生交联反应,最终形成体型(三维网状)结构的丙阶树脂。固化后不可熔化、不可溶解(热固性)。
酸催化体系(Novolac / 线性树脂)
反应条件:苯酚过量(苯酚 : 甲醛摩尔比通常为 1 : 0.8),催化剂为酸(如盐酸、硫酸)。
反应机理:
- 酸催化剂质子化甲醛的羰基氧,使其转化为强亲电试剂(碳正离子 \ce{^+CH2OH})。
- 碳正离子攻击苯酚的邻 / 对位,生成羟甲基苯酚中间体。
- 羟甲基在酸性条件下极不稳定,会迅速被质子化并脱去一分子水,生成高度活泼的苄基碳正离子(\ce{HO-C6H4-CH2+})。该碳正离子立刻攻击另一分子苯酚的邻 / 对位,形成以亚甲基桥连接的二聚体。
- 反应不断重复,生成由亚甲基桥连接的线性或支链聚合物。
固化:线性酚醛树脂自身加热无法固化(因为没有交联基团),必须加入交联剂。工业上最常用的是六亚甲基四胺(HMTA,俗称乌洛托品)。在加热时,HMTA 分解提供甲醛和氨,充当交联桥梁和催化剂,最终交联成三维网状网络。
性能与应用
酚醛树脂拥有高热稳定性、阻燃、高机械强度、低成本和优异的电绝缘性,在今天依然具有不可替代的地位:
- 胶粘剂与木材工业:制造室外用胶合板、定向刨花板(OSB)。
- 模塑料与电器件:锅把手、开关面板、电表壳等,利用其绝缘、耐热、不燃烧的特点。
- 复合材料与电子工业:浸渍纸或玻璃布后压制成层压板,覆铜板(CCL)的基材很多就是酚醛树脂做的。
- 摩擦材料:汽车刹车片、离合器摩擦片的粘结剂。
- 航空航天烧蚀材料:火箭发动机喷管、飞船重返大气层的防热罩。
聚乙烯醇
聚乙烯醇(Polyvinyl Alcohol,简称 PVA)是一种具有独特合成路径的水溶性高分子,化学式为 [\ce{CH2CH(OH)}]_n。
为什么不能直接聚合
乙烯醇(\ce{CH2=CH-OH})是一种极不稳定的烯醇。根据烯醇 - 酮互变异构原理,乙烯醇会迅速自发地异构化为更稳定的乙醛(\ce{CH3CHO}):
\ce{CH2=CH-OH <=> CH3CHO}
因为单体无法稳定存在,所以不能通过常规加聚反应来合成 PVA。
间接合成法(工业两步法)
工业上合成 PVA 采用「先聚合,后水解(醇解)」的间接路线:
第一步:合成聚醋酸乙烯酯(PVAc)。单体选用醋酸乙烯酯(Vinyl acetate, VAc),发生自由基聚合,生成聚醋酸乙烯酯:
n\;\ce{CH2=CH(OCOCH3)} \xrightarrow{\text{引发剂}} [\ce{-CH2-CH(OCOCH3)-}]_n
第二步:聚醋酸乙烯酯的醇解(或水解)。将 PVAc 溶解在甲醇中,加入碱(如氢氧化钠)作为催化剂,发生酯交换反应(醇解),将侧链的醋酸酯基替换为羟基:
[\ce{-CH2-CH(OCOCH3)-}]_n + n\;\ce{CH3OH} \xrightarrow[\ce{CH3OH}]{\ce{NaOH}} [\ce{-CH2-CH(OH)-}]_n + n\;\ce{CH3COOCH3}
醇解机理
工业上通常采用碱催化的甲醇醇解法,这是经典的亲核酰基取代机理:
- 氢氧化钠与甲醇反应,生成甲氧基负离子(\ce{CH3O-})。
- \ce{CH3O-} 攻击 PVAc 侧链酯基的羰基碳,形成四面体中间体。
- 四面体中间体碳氧键断裂,释放出醋酸甲酯。
- 高分子侧链的氧负离子从甲醇中夺取质子,生成羟基(-OH),同时再生 \ce{CH3O-} 继续催化反应。
醇解的程度决定了 PVA 的规格。完全将酯基替换为羟基称为完全醇解(纯度 >98\%);若保留部分酯基,则称为部分醇解(如 87\% \sim 89\% 醇解度)。
性质
PVA 的性能高度依赖于它的聚合度(分子量大小)和醇解度(羟基的比例)。
- 完全醇解的 PVA:只有在热水(\pu{90^oC} 以上)中才能溶解,冷水中不溶。原因是分子链上全都是 -OH,分子内和分子间形成了极强的氢键,导致结晶度很高,冷水无法破坏其晶格。
- 部分醇解的 PVA(如 88\%):反而在冷水中溶解度很好。因为主链上残留的体积较大的醋酸酯基破坏了氢键的规律性,降低了结晶度,水分子更容易渗入。
化学改性
PVA 主链上密布着仲羟基(-OH),这使其具有类似多元醇的化学性质:
- 缩醛化反应:PVA 与醛类在酸性催化剂下反应生成缩醛。例如聚乙烯醇缩甲醛(PVF)用于制造维纶纤维;聚乙烯醇缩丁醛(PVB)具有极好的透明度和韧性,用于汽车挡风玻璃的夹层防爆膜。
- 交联反应:硼酸根离子能与 PVA 链上的羟基形成动态共价键,将线型高分子交联成网状结构(常见的「史莱姆」玩具就是利用这一原理)。
- 共聚拓展:在聚合时引入乙烯,醇解后得到乙烯 - 乙烯醇共聚物(EVOH),是目前阻隔氧气性能最好的包装材料之一。
应用
- 水溶性薄膜与包装:洗衣凝珠的外包装膜、农药包装袋。
- 纺织与纤维:缩醛化后制成维纶,吸湿性好,最接近棉花的合成纤维。
- 粘合剂与建筑材料:纸张、木材的粘合剂,水泥的增稠剂。
- 生物医学工程:PVA 水凝胶用于制造人工软骨、隐形眼镜、医用敷料、人工泪液。
- 聚合分散剂:合成聚氯乙烯(PVC)或聚苯乙烯(PS)时不可或缺的高分子乳化剂。
碳骨架构建
碳链延长
炔醛酮与 \ce{HCN} 的加成反应:
\ce{\underset{乙炔}{HC#HC} ->[HCN] \underset{丙烯腈}{H2C=CH-CN} ->[H2O(H+)][\triangle] CH2=CH-COOH}
\ce{(CH3)2C=O ->[HCN] (CH3)2CH-CN ->[H2O(H+)][\triangle] (CH3)2CH-COOH}
\ce{CH3CHO ->[HCN] CH3(OH)-CN ->[H2] CH3OH-CH2NH2}
炔烃和醛发生加成反应:
\ce{R-CH#CH2 + R'CHO -> R-C#C-CH(OH)-R'}
卤代烃与氰化钠:
\ce{CH3CH2Br ->[NaCN] CH3CH2CN ->[H2O(H+)] CH3CH2COOH}
羰醛缩合反应:
羰基化合物分子中在羰基邻位碳原子上的氢原子(\alpha-\ce{H})受羰基吸引电子作用的影响,具有一定的活泼性,分子内含有 \alpha-\ce{H} 的醛在一定条件下可以发生加成反应,生成 \beta-羟基醛,该产物易失水,得到 \alpha,\beta-不饱和醛。

乙醛拥有三个,因此可以发生三次羟醛缩合反应。
部分信息题会跳过羟基醛的步骤,直接得到不饱和醛。
格氏试剂:
卤代烃可以与金属反应,其中最负盛名的是有机镁化合物,它是由法国化学家格利雅发现。通过卤代烃 \ce{RX} 与镁(用醚作溶剂)作用得到烃基卤化镁 \ce{RMgX},也称为格氏试剂。
烃基卤化镁与其他物质(如卤代烃、醛、二氧化碳等)反应可以实现碳链增长,得到烃、醇、羧酸、酮等多种有机化合物。
\ce{CH3CH2MgBr} \begin{cases} \ce{->[ROH or H2O] &CH3CH3}\\ \ce{->[CH3Br] &CH3CH2CH3}\\ \ce{->[CO2 or H2O/H+] &CH3CH2COOH}\\ \ce{->[HCHO or H2O/H+] &CH3CH2CH2OH} \end{cases}
卤代烃与醇钠反应实现不对称醚的合成:
这是制备不对称醚的一种常用方法(当然也可以合成对称醚),被称为威廉森合成法。
\ce{RX + R'ONa -> ROR' + NaX}
也就是卤代烃在氢氧化钠醇溶液中的醇解。
烷基化反应(傅列德尔-克拉夫茨反应):
芳烃在无水 \ce{AlCl3} 等路易斯酸的催化下,苯环上的氢被烷基取代的反应:
\ce{C6H6 + R-Cl ->[AlCl3] C6H5R + HCl}
反应机理如下:
\ce{R-Cl + AlCl3 -> R+ + AlCl4-}
\ce{C6H6 + R+ -> C6H5R + H+}
在烷基化反应中,进攻苯环的亲电试剂为烷基碳正离子。
在催化剂作用下产生碳正离子的化合物,如卤代烃、烯烃、环氧乙烷和醇均可作为烷基化试剂。\ce{AlCl3} 是烷基化反应中活性最高的催化剂,此外,\ce{FeCl3}、\ce{ZnCl2}、\ce{HF}、\ce{H2SO4} 也可作为催化剂。
碳链缩短
高锰酸钾氧化:
氧化醇。
氧化醛和酮。
氧化烯烃、炔烃。
氧化芳香烃(如甲苯)。
酯的水解:
酸性水解:生成酸和醇。
碱性水解:生成盐和醇。
成环反应
狄尔斯-阿尔德反应:
共轭二烯烃与含碳碳双键(或三键)的化合物在一定条件下反应,得到环加成产物,构建了环状碳骨架,该反应用于构建六元碳环骨架。

理解:共轭二烯烃(如 1,3-丁二烯)的两根双键各自打开一半,与含亲双烯体(在这里是丙烯酸)的碳碳双键发生加成反应,两个“半截键”与原来的单键共同形成了新的双键,得到了环加成产物,构建了环状碳骨架。
酯化成环:如二元酸与二元醇的酯化反应、羟基酸的酯化反应。
二元醇成环:
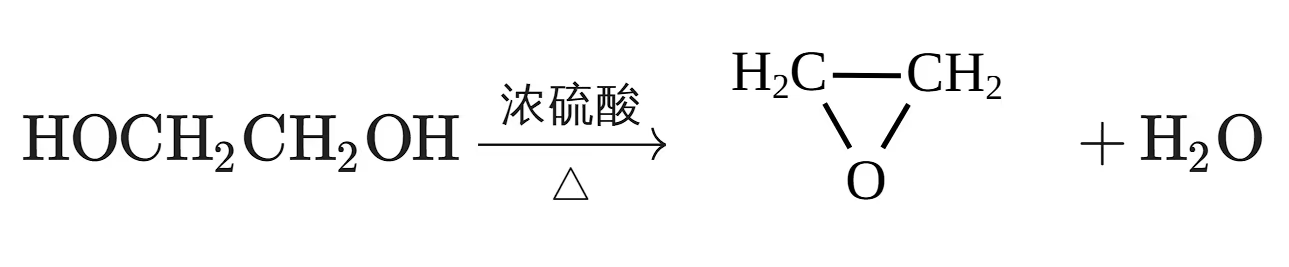
二元羧酸成环:
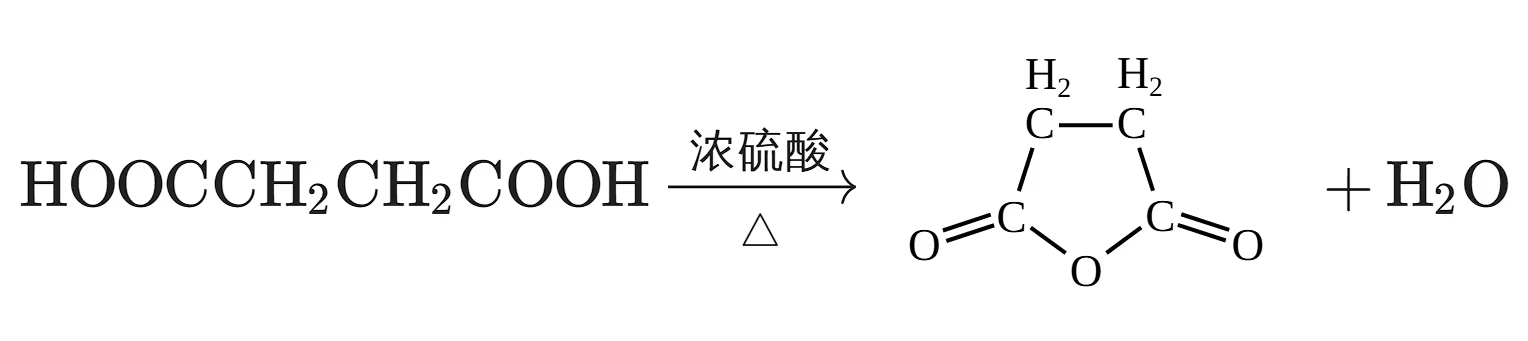
氨基酸成环:
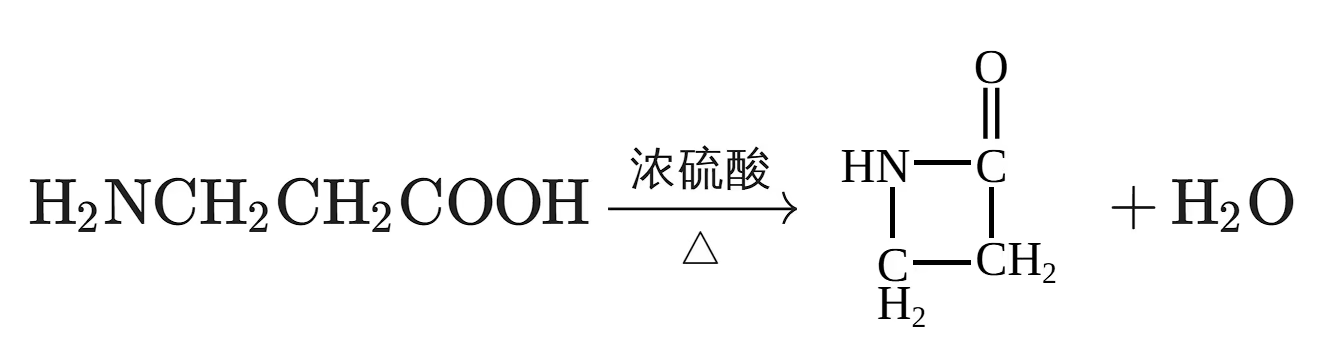
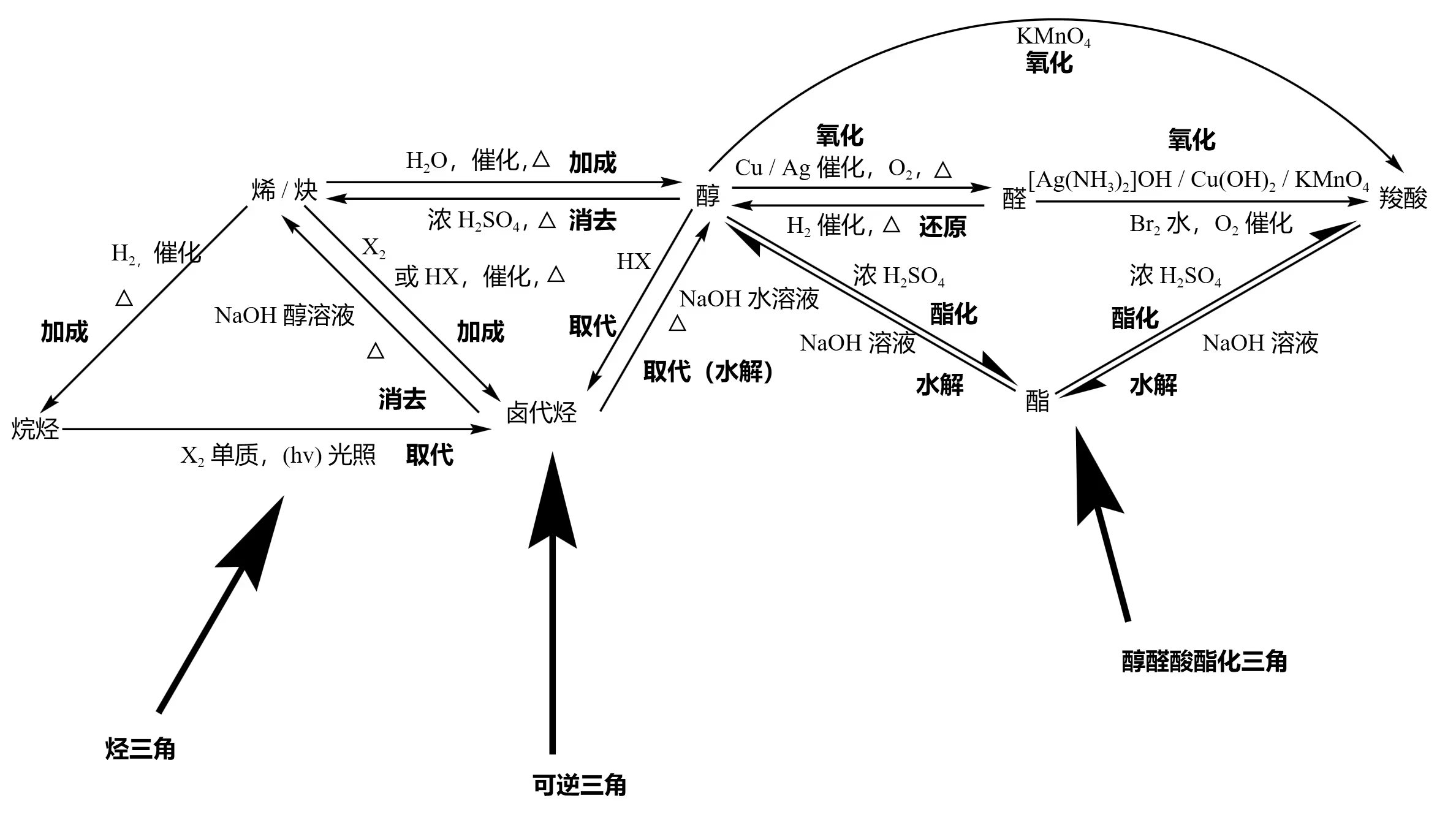
官能团的转化
引入官能团
引入碳碳双键:
卤代烃的消去反应。
\ce{CH3CH2Br + NaOH ->[乙醇][\triangle] CH2=CH2 ^ + NaBr + H2O}
醇的消去反应。
\ce{CH3CH2OH ->[浓硫酸][\pu{170^oC}] CH2=CH2 ^ + H2O}
引入碳卤键:
烃、酚的取代反应。
不饱和烃、醇与 \ce{X2},\ce{HX} 的取代反应。
引入羟基:
烯烃与水的加成反应。
醛、酮与氢气的加成。
卤代烃在碱性条件下水解。
\ce{CH3CH2Br + NaOH ->[H2O][\triangle] CH3CH2OH + NaBr}
酯的水解。
通过硼氢化钠引入羟基或醛基。
\ce{CH3CHO ->[NaBH4] CH3CH2OH}
引入醛基或羰基:
醇的催化氧化。
\ce{2CH3CH2OH + O2 ->[Cu/Ag][\triangle] 2CH3CHO + 2H2O}
含碳碳三键的物质与水加成。
\ce{HC#CH + H2O ->[催化剂][\triangle] H2C=CHOH ->[异构化] CH3-CHO}
碳碳双键氧化:叔碳变羰基。
引入羧基:
醇、醛的氧化:酸性高锰酸钾,铜、银镜反应、斐林试剂。
碳碳双键的氧化:
\ce{R-CH=CH-R' ->[KMnO4][H+] R-COOH + R'COOH}
酯、酰胺键的水解:酸去羟基醇 / 胺去氢。
引入酯基:酯化反应、酰基化反应。
通过重氮盐引入多种官能团:芳香族伯胺(\ce{Ar-NH2})在强酸(\ce{HCl} 或 \ce{H2SO4})和亚硝酸钠(\ce{NaNO2})的作用下,于 0 \sim 5^{\circ}\ce{C} 冰水浴中生成重氮盐(\ce{Ar-N2^+X^-})。重氮基是一个极佳的离去基团(放出 \ce{N2}),可以被丰富的亲核试剂取代,从而在芳环上引入各类官能团。
\ce{Ar-NH2 ->[NaNO2, HCl][0\sim5^\circ C] Ar-N2^+Cl^-}
主要转化途径包括:
Sandmeyer 反应:与 \ce{CuCl}、\ce{CuBr}、\ce{CuCN} 反应,分别引入 -\ce{Cl}、-\ce{Br}、-\ce{CN}。
Schiemann 反应:与 \ce{HBF4} 反应生成沉淀,加热分解后引入 -\ce{F}(芳氟化合物极难通过直接氟化合成)。
水解反应:加水加热,转化为酚(-\ce{OH})。
碘化反应:加 \ce{KI}(无需铜催化),引入 -\ce{I}。
脱氨基反应:加次磷酸(\ce{H3PO2})或乙醇,重氮基被 -\ce{H} 取代,实现氨基的定向去除。
偶合反应:与富电子芳环(酚、胺)反应,生成偶氮化合物(-\ce{N=N}-),是合成偶氮染料的核心反应。
定位效应的切换策略:利用硝基(间位定位)和氨基(邻对位定位)定位方向的差异,在芳环不同位置分步引入取代基。典型路线为“硝化 \rightarrow 还原 \rightarrow 邻对位取代”。
例如合成对溴苯胺:
\ce{苯 ->[HNO3/H2SO4] 硝基苯 ->[Fe/HCl] 苯胺 ->[(CH3CO)2O] 乙酰苯胺 ->[Br2] 对溴乙酰苯胺 ->[水解] 对溴苯胺}
先利用硝基的间位定位完成第一步转化,还原为氨基后切换为邻对位定位,再引入其他基团。
无痕导向基策略:当需要在特定位置引入基团但常规定位规律不允许时,可以先引入氨基作为“占位基团”,利用其强大的邻对位活化能力将目标基团引导至指定位置,再通过重氮化 + 脱氨基反应将氨基去除。
例如合成 1,3,5-三溴苯:苯胺直接溴化生成 2,4,6-三溴苯胺(氨基的强活化使三个邻对位同时被取代),经重氮化后用 \ce{H3PO2} 脱除重氮基,即可得到 1,3,5-三溴苯。氨基在此策略中扮演了“活化、定位、事后消失”的角色。
改变官能团
消除官能团:
- 通过加成反应消除不饱和键。
- 通过消去、氧化、酯化反应消除羟基。
- 通过加成、氧化反应消除醛基。
- 通过水解反应消去酯基、酰胺基、碳卤键。
改变官能团的种类:
改变官能团的数目:
\begin{aligned} \ce{ CH3CH2OH &->[消去][-H2O] CH2=CH2 \\ &->[加成][+Cl2] Cl-CH2-CH2-Cl \\ &->[水解] HO-CH2-CH2-OH } \end{aligned}
改变目标官能团的物质:通过不对称烯烃与卤化氢的加成改变官能团的位置(运用马氏规则)。
\begin{aligned} \ce{ CH3CH2CH2Cl &->[消去][-HCl] CH3CH=CH2 \\ &->[加成][+HCl] CH3-CH(Cl)-CH3} \end{aligned}
保护官能团
含有多个官能团的有机化合物在进行反应时,非目标官能团也可能受到影响,此时需要将受影响的官能团保护起来,先将其转化为不受该反 应影响的其他官能团,反应后再将受影响的官能团复原。
羟基的保护:合成反应会影响羟基,无法直接转化。
\ce{R-OH ->[保护基] R-O-R' ->[\text{合成反应}] R''-O-R' ->[脱保护基] R''-OH}
如果体系受强碱影响,就用成醚反应保护,如果要受氧化,就要用酯化反应保护。
如果是酚羟基,也可以先用氢氧化钠变为 \ce{-ONa},然后再酸化还原出酚羟基。
碳碳双键的保护:
碳碳双键易被氧化,在氧化其他基团前,可通过与卤素单质、卤化氢等加成的方法先将碳碳双键保护起来,待氧化其他基团后,再通过消去反应(\ce{NaOH} 醇溶液,加热)重新转化为碳碳双键。
羧基:羧基遇到高温容易脱羧。
氨基的保护:游离氨基(-\ce{NH2})非常活泼,直接卤化会导致多卤代(如苯胺加溴水直接生成 2,4,6-三溴苯胺),且在硝化等氧化性条件下会被破坏。通过酰化反应(如加乙酸酐或乙酰氯)将 -\ce{NH2} 转化为酰胺基 -\ce{NHCOCH3},既降低了芳环活化程度(控制取代产物为单一取代),又保护氨基不被氧化。反应完成后,再通过酸或碱水解恢复氨基。
\ce{Ar-NH2 ->[(CH3CO)2O] Ar-NHCOCH3 ->[\text{合成反应}] Ar'(NHCOCH3) ->[H2O/H+ \text{or} OH-] Ar'-NH2}
油脂的工业提取
浸出法与压榨法
油脂的工业提取主要有两种工艺:压榨法和浸出法。
压榨法是通过机械外力直接对油料作物施加巨大压力,将油脂从原料中挤压出来。这种方法类似于挤海绵,保留了油料原有的风味和部分营养成分,但出油率较低,压榨后的饼粕中仍会残留约 7\% \sim 9\% 的油脂,导致生产成本较高。
浸出法则是利用有机溶剂进行油脂提取的化学方法。生产中使用的溶剂并非日常所说的汽油,而是一种专门生产的食品级六号溶剂油,其主要成分为正己烷(\ce{C6H14})。由于正己烷的沸点较低(约 60^{\circ}\ce{C} \sim 70^{\circ}\ce{C}),而油脂的沸点高达 200^{\circ}\ce{C} 以上,因此可以通过蒸馏的方式将溶剂与油脂分离。
浸出法的工艺流程如下:首先将预处理后的油料碎料(称为“料坯”)浸泡在正己烷中,利用“相似相溶”原理,油脂会溶解在溶剂中形成“混合油”;随后通过加热蒸发,沸点低的正己烷变成气体逸出并被回收循环使用,剩下的即为浸出毛油;最后经过脱胶、脱酸、脱色、脱臭等一系列精炼工序,得到最终的食用油。
浸出法的最大优势在于出油率高,可将饼粕中的残油率降至 1\% 以下,极大提高了油脂的利用率,降低了食用油的生产成本。目前全球大宗食用油(如大豆油、玉米油、菜籽油等)普遍采用浸出法生产。
溶剂残留与安全性
正规厂家生产的浸出油是安全可食用的。中国国家标准对食用油中的溶剂残留有严格规定:一级和二级食用油的溶剂残留必须“不得检出”;三级和四级食用油允许的残留量不得超过 \pu{50mg/kg},这一微量水平对人体完全无害。在高温烹调过程中,极其微量的残留也会瞬间挥发。
压榨油与浸出油的选择
| 特性 | 浸出油 | 压榨油 |
|---|---|---|
| 工艺 | 化学溶剂(正己烷)提取 | 物理机械压榨 |
| 出油率 | 高,经济高效 | 低 |
| 价格 | 相对便宜 | 相对昂贵 |
| 风味 | 精炼后纯净、无味 | 保留原料原有风味 |
| 安全性 | 合格产品完全安全 | 无溶剂残留风险 |
消费者可根据需求选择:追求经济实惠和烟点高的可以选择浸出油;追求天然风味的可以选择压榨油。购买时应查看产品标签上的工艺标识(压榨/浸出)和质量等级。
医用黏合剂
概述
医用黏合剂是现代医学中用于替代或辅助传统缝合线、吻合器,实现组织粘接、止血、封闭创面或固定医疗器械的重要材料。其发展是现代材料科学、有机化学与临床医学交叉融合的典范。其核心挑战在于:在湿润、动态、复杂且敏感的生理环境中,实现快速、牢固且安全的黏合。
氰基丙烯酸酯类黏合剂
氰基丙烯酸酯类是目前临床和日常生活中最常见的强力黏合剂。
化学结构:通式为 \ce{CH2=C(CN)COOR}。双键碳上同时连有两个强吸电子基团:氰基(\ce{-CN})和酯基(\ce{-COOR}),使双键的电子云密度大大降低,极易受到亲核攻击。
聚合固化机理:
阴离子聚合:在组织表面微量的水或弱碱性物质(如氨基酸、蛋白质的氨基)的引发下,双键迅速打开,发生链式增长,瞬间(数秒内)形成高分子量的聚氰基丙烯酸酯长链。
\ce{n CH2=C(CN)COOR ->[-\ce{H2O}][\ce{NH2}] \poly{CH2-C(CN)(COOR)}}
这是一个放热反应。固化过程释放的热量是临床关注点之一(可能引起组织热损伤)。
结构修饰与性能关系:
- 短链酯(如甲酯、乙酯):聚合极快,固化后脆性大。降解产物含有较高浓度的甲醛,对组织有毒性,不能用于医用内部组织。
- 长链酯(如正丁酯、辛酯):如临床常用的氰基丙烯酸辛酯。长烷基链的引入增加了分子的柔韧性,使得胶体固化后能跟随皮肤拉伸;同时降解速度减慢,甲醛释放率低于组织的耐受阈值,因此具有极佳的生物相容性。
纤维蛋白黏合剂
纤维蛋白黏合剂是模拟人体凝血最后阶段的自源性生物黏合剂。
化学与生化组成:主要成分来自人血浆。
- 组分 A:高浓度的纤维蛋白原、凝血因子 XIII(纤维蛋白稳定因子)。
- 组分 B:凝血酶、氯化钙。
固化机理:
酶促反应:凝血酶将纤维蛋白原水解,切下小分子的纤维蛋白肽 A 和 B,暴露出结合位点,形成可溶性的纤维蛋白单体。
自组装:纤维蛋白单体自发聚合并交联,形成不溶性的纤维蛋白多聚体网络(软凝块)。
共价交联:在 \ce{Ca^{2+}} 存在下,凝血因子 XIII(一种转谷氨酰胺酶)被凝血酶激活。激活的 FXIIIa 催化纤维蛋白链间 \varepsilon-(\gamma-谷氨酰) 赖氨酸异肽键的形成,使凝胶网络共价交联,强度大幅提高。
\ce{\text{谷氨酰胺残基} + \text{赖氨酸残基} ->[FXIIIa][\ce{Ca^{2+}}] \text{共价交联} + \ce{NH3}}
聚氨酯类黏合剂
聚氨酯类黏合剂是一类具有高弹性的组织密封剂。
化学结构:含有异氰酸酯基团(\ce{-N=C=O})的预聚物。
反应机制:
- 与组织键合:\ce{-NCO} 基团高度活泼,可与组织蛋白上的氨基(\ce{-NH2})发生加成反应生成脲键(\ce{-NH-CO-NH-}),或与羟基(\ce{-OH})反应生成氨基甲酸酯键(\ce{-O-CO-NH-}),形成牢固的共价键。
- 自我交联:\ce{-NCO} 遇组织水分发生水解,生成不稳定的氨基甲酸,随即脱羧放出二氧化碳(\ce{CO2}),生成伯胺。伯胺再与未反应的 \ce{-NCO} 反应,形成交联网络。
通过调节预聚物中多元醇的链长和亲水性,可以精准调控聚氨酯胶的弹性和降解时间,尤其适合心脏血管等需要高弹性的跳动组织。
贻贝仿生黏合剂
贻贝能在狂风巨浪的海水中紧紧吸附在礁石上,其核心在于其足丝蛋白中富含的特殊氨基酸。
关键化学基团:邻苯二酚(或称多巴)。其在碱性或氧化条件下可氧化为邻苯醌。
黏附机理:
- 配位键:邻苯二酚的酚羟基可与组织表面的金属离子(如 \ce{Fe^{3+}})或金属氧化物形成强配位键。
- 氢键与疏水相互作用:与各种表面广泛存在。
- 共价键:邻苯醌可与蛋白质的巯基(\ce{-SH})或氨基(\ce{-NH2})发生迈克尔加成或席夫碱反应,形成共价连接。
贻贝仿生黏合剂实现了在湿润、盐水环境下的强黏附,是医用胶领域的重要仿生突破。
聚乙二醇基黏合剂
聚乙二醇(PEG)基黏合剂是通过化学修饰,创造可与组织形成共价键的“分子胶”。
代表性体系:
PEG-琥珀酰亚胺酯:末端活化酯基团可与组织蛋白质的伯氨基(主要来自赖氨酸残基)发生高效的酰胺化反应,形成共价键。
\ce{\text{PEG}-\ce{COO-NHS} + \ce{H2N}-\text{Protein} -> \text{PEG}-\ce{CONH}-\text{Protein} + \ce{NHS}}
席夫碱反应体系:如氧化海藻酸钠(含醛基)与明胶(含氨基)混合。醛基与氨基反应形成动态共价的亚胺键(\ce{C=N}),凝胶化温和、可调。
优势:黏合强度高(共价键)、生物相容性好、可功能化(如载药)。
生物降解与生物相容性
生物降解途径:
- 水解:聚酯类、氰基丙烯酸酯类的骨架容易受到水分子的攻击发生断裂。
- 酶促降解:生物来源的胶(如纤维蛋白胶)会被体内的纤溶酶和蛋白酶识别并切割成氨基酸。
- 巨噬细胞吞噬:降解的微小碎片会被免疫细胞(巨噬细胞)吞噬,随后进入代谢循环。
毒理与异物反应:
- 氰基丙烯酸酯降解产生甲醛,若浓度超过线粒体代谢能力,会导致局部细胞坏死。
- 任何人工合成胶进入体内都会被免疫系统识别为“非己”。轻度的急性炎症有助于招募成纤维细胞促进愈合;但若化学交联剂毒性大,会导致慢性炎症,最终形成厚厚的纤维囊包裹,阻碍组织再生。
临床应用
不同化学特性的医用黏合剂对应不同的临床场景。
氰基丙烯酸酯(尤其辛酯):
- 表皮切口闭合:替代缝线,美观、无需拆线、防水。
- 紧急止血:肝、脾等实质性脏器创面的快速封闭。
- 血管栓塞:介入治疗中栓塞动静脉畸形。
纤维蛋白胶:
- 组织贴合与密封:吻合口加固、防止肺泡漏气、硬脊膜封闭。
- 止血:用于渗血创面,特别适用于凝血功能障碍患者。
- 药物/细胞载体:作为生长因子或干细胞缓释支架。
聚氨酯与 PEG 基黏合剂:
- 高强度需求部位:软骨修复、角膜穿孔封堵。
- 微创手术:内镜下递送,原位凝胶化封闭溃疡或瘘管。
煤油的工业制备
石油分馏
煤油并非通过“合成”获得,而是主要通过物理分离和化学精制从天然原油中得到。
核心原理:分馏(蒸馏)。利用原油中各组分的沸点不同进行分离。煤油是沸点范围约为 150^{\circ}\ce{C} \sim 250^{\circ}\ce{C} 的石油馏分,主要成分为 C_{11} \sim C_{16} 的烷烃、环烷烃和芳香烃。
主要步骤:
- 原油预处理:脱盐、脱水。
- 常压分馏:将加热后的原油送入分馏塔,在塔的中上部(介于汽油和柴油馏分之间)收集煤油馏分。
- 精制与脱硫:加氢精制是目前最主要的方法。在催化剂作用下,通入氢气,脱除硫、氮、氧等杂质,并使不饱和烃饱和,能显著降低硫含量,减少环境污染。
煤的液化
在我国“富煤贫油”的国情下,通过煤的液化合成煤油是重要的能源补充途径。
间接液化工艺:
煤气化:生成合成气(水煤气)
\ce{C + H2O(g) ->[高温] CO + H2}
费托合成:在催化剂作用下,合成气转化为液态烃
\ce{CO + H2 ->[催化剂][加热加压] 液态烃(煤油等) + H2O}
STSE 视角
主要用途:航空煤油(喷气燃料)、灯用煤油、炉用燃料、溶剂等。航空煤油是现代航空运输业的“血液”,关乎交通与经济命脉。
环境与影响:
- 正面:相比直接燃烧原油,分馏利用效率更高。
- 挑战:原油开采和炼制过程存在污染风险;未经精制的煤油燃烧会产生含硫化合物(如 \ce{SO2}),导致酸雨;最终燃烧产物(\ce{CO2})是温室气体。
- 应对:通过加氢脱硫等清洁精制技术降低产品硫含量;发展新能源作为长远替代。
应试要点总结:
- 核心考法:以煤油为例,考查石油分馏的原理、馏分获取方式和绿色精制技术(加氢)。
- 关键答案:
- 来源:石油(原油)。
- 方法:分馏(蒸馏),属于物理变化;煤的液化属于化学变化。
- 精制关键:加氢脱硫(加氢精制),属于化学变化。
- STSE 视角:联系能源利用、环境污染与防治(酸雨)、绿色化学工艺。
简单记忆:煤油是从石油里“分”出来的,也可以通过煤的液化“合”成,经过加氢精制后才能清洁使用。